SUMMARY
Part two of two. This is the second part of a work about drug metabolism and its clinical importance. In this part will be explained drug interactions of outstanding interest. Moreover the mechanisms of the interaction will be described in order to show the importance of being informes of the metabolism of medicaments.
INTRODUCCIÓN
Esta es la segunda parte de una serie de dos artículos sobre el tema del metabolismo de medicamentos. En esta sección, después de haber explicado las bases del metabolismo citocromal de los medicamentos en la primera parte de esta revisión, se abordarán diferentes ejemplos en los cuales están involucrados medicamentos de uso cotidiano. Los ejemplos que se mencionarán revisten una especial importancia pues las interacciones que se comentan son de gran interés en la práctica médica diaria. Se trata de no hacer un aburrido resumen de las interacciones, sino más bién de presentar aquellas cuyo mecanismo es conocido y permite una explicación sobre una base científica adecuada. Se incluyen interacciones con antibióticos tipo macrólidos, antifúngicos azólicos, antidepresivos tipo inhibidores de la recaptura de serotonina, antiepilépticos y anticonceptivos orales. Las interacciones son prevenibles si se conocen y hasta predecibles si se entiende su mecanismo.
Palabras clave: citocromos, macrólidos, antifúngicos, antiepilépticos interacciones, anticonceptivos.
ANTICONCEPTIVOS HORMONALES ORALES
Sangrados intermenstruales y aumento de los embarazos han si observados al combinar anticonceptivos con fármacos inductor enzimáticos. El 17 a-etinilestradiol es metabolizado por el CYP 3A4 a través de una (2-) hidroxilación (19). A este tipo de metabolismo está sujeto cerca de un 30% del estrógeno ingerido por los humanos (20), y puede ser acelerado por la inducción enzimática que causa la rifampicina, los antiepilépticos: fenobarbital, fenitoína, primidona, carbamazepina y el antifúngico griseofulvina (26,5). La rifampicina (60mg/día) por 6-10 días duplica y hasta triplica el citocromo hepático, sobre todo el CYP3A4 (20). Se recomienda que mujeres tomando antiepilépticos deberían consumir anticonceptivos orales con cerca de 50 mg de etinilestradiol. La Familiy Planning Association de Inglaterra recomienda el uso de anticonceptivos monofásicos por tres meses sin interrupción, después de lo cual se descansa un corto período de cuatro días (25). También hay que tener precaución al combinar otros estrógenos como levonorgestrel y la noretindrona con inductores enzimáticos (4).
De los antiepilépticos
más recientes la oxcarbazepina (21) y el topiramato
(23) pueden inducir y afectar los niveles de anticonceptivos
orales. Por otro lado es conocido que anticonceptivos con bajas dosis de
estrógenos «50mg) pueden reducir el metabolismo microsómico
de la cafeína por interferencia con el CYP 1A2 (1),
da tal manera que tendrían un efecto inhibidor sobre este citocromo.
El progestágeno gestodene, presente en algunas combinaciones de
anticonceptivos: Harmonet", Gynovin", Femiane" y Minulet", es un inhibidor
del CYP 3A4 y se creé que es el responsable del aumento de cortisol
y etinilestradiol que se observa en mujeres tomando esta combinación
de anticonceptivos (19).
| CATEGORIA TERAPEUTICA MEDICAMENTO | |
| Antiepilépticos | Carbamazepina, fenitoína etosuximida, topiramato, lamotrigina, valproato |
| Antiinfecciosos | ltraconazol, mebendazol |
| Bloqueadores de canales de calcio | Nifedipina, verapamil, fenlodipina, diltiazem |
| Agentes cardiovasculares | Quinidina, digoxina propranolol, procainamida |
| Corticosteroides | Dexametasona, prednisolona,
pirednisona, metilprednisolona |
| Anticoagulantes orales | warfarina |
| Anticonceptivos orales y hormonas sexuales | Estrógenos conjugados etinilestradiol, levonorgestrel, noretindrona |
MACRÓLIDOS Y ANTIFÚNGICOS AZÓLICOS
Tanto el antifúngico
imidazólico ketoconazol, como los triazoles: itraconazol y fluconazol
son conocidos inhibidores del CYP 3A4. Además, el fluconazol es
un inhibidor del CYP2C9 (10,24,17).
El fluconazol tiene un efecto intermedio de inhibición comparado
con el ketoconazol, que es el más fuerte, y el itraconazol que es
el más débil. El miconazol, en los países que hay
presentación oral, se le considera un inhibidor moderado del CYP3A4
(24) y puede interaccionar con la warfarina y ciclosporina
disminuyendo su metabolismo tal y como lo hace el ketoconazol, itraconazol
y fluconazol(23). El fluconazol se comporta, in vitro,
como un débil inhibidor de los citocromos, sin embargo, alcanza
altas concentraciones en el hígado por lo que sus efectos podrían
ser mayores de los estimados (35,18).
El fluconazol a dosis de 200mg/día aumenta el AVC de la fenitoína
en un 75% (6). Esta interacción sería de
importancia, sobre todo, cuando se utilicen dosis altas de fluconazol como
las que se emplean en el tratamiento de la candidiasis sistémica
o la meningitis por criptococos y a la vez el paciente este con tratamiento
con fenitoína. Dicha interacción podría ser el resultado
del bloqueo del CYP2C9 por parte del fluconazol, ya que la fenitoína
es metabolizada por este citocromo. Además, el fluconazol presenta
interacciones con la carbamazepina, sulfametoxazol e hipoglicemiantes orales
tipo sulfonilureas (23). El itraconazol disminuye el
aclaramiento de digoxina y aumenta sus niveles plasmáticos (2).
|
JAPONESES
20%
TABLA #2. INCIDENCIA DE
POBRES
|
La terbinafina (lamisil"),
antifúngico más reciente, recomendado para micosis superficiales
y que pertenece al grupo de las alilaminas parece tener débiles
efectos inhibidores enzimáticos. Dosis única de 500 mg de
terbinafina no alteraron las concentraciones de cortisol, testosterona,
prolactina, 17-OH progesterona y de la hormona folículo estimulante
en plasma, indicando que no hay inhibición de los citocromos responsables
de metabolizar estas hormonas (19). En vitro si se ha
observado una inhibición del metabolismo de la ciclosporina (12%)
y del etinil estradiol (35%). Estudios en voluntarios han mostrado
una reducción del aclaramiento de la cafeína, indicando una
posible interacción con el CYP 1A2 (32). Los efectos
clínicos de estas últimas interacciones no están claros.
La cisaprida es una benzamida sustituida con actividad procinética
de uso en Costa Rica. Se sabe que este fármaco despliega sus efectos
al estimular receptores 5-HT4 en el plexo mientérico, tanto en el
tracto gastrointestinal alto como en el bajo. Posee escaso efecto antiemético
(14). La cisaprida puede producir síncope, prolongación
del QTc y torsades de pointes, sobre todo cuando se sobredosifica, existe
una arritmia previa o se están consumiendo antiarrítmicos
(35,14). Esta acción de la cisaprida
podría ser el resultado de la estimulación directa de receptores
5-HT4 en los atrios del corazón, donde median el aumento en la fuerza
de contracción (22). Al ser la cisaprida metabolizada
por el CYP 3A4 se pueden generar arritmias potencialmente mortales cuando
se le combina con inhibidores de este citocromo, a saber: azoles (ketoconazol,
fluconazol itraconazol, metronidazol) o macrólidos (eritromicina
y claritromicina) (30,35,14).
Hay reporte que mencionan la posibilidad de que la eritromicina por si
misma produzca torsade de pointes (16).
|
|
|
|
| Inicio de efecto | 8-12 h. | 3-6 h. |
| Efecto máximo | 3-5 días | 24-48 h. |
| Duración del efecto | 5-7 días | 5-12 días |
| Peso del hígado | Aumento | Poco aumento |
| Retículo endoplasmático | Aumento | Poco cambio |
| Flujo sanguíneo hepático. | Aumento | Sin cambio |
| Flujo biliar | Aumentado | Sin cambio |
| Citocromo P-450 | Aumentado CYP 2B1 | Aumento CYP1A1 y CYP 1A2 |
| Epoxidohidrolasa | Aumento | Poco cambio |
| Glucuronidisación
(UGT2B1) |
Aumento | Poco cambio |
| Glutatión transferasa | GST-1 | GST 3-4 |
| NAD-menadion-óxido
reductasa |
Sin cambio | Aumento |
| NADPH-citocromo
reductasa |
Aumento | Sin cambio |
Tampoco se recomienda la administración conjunta de azoles (ketoconazol, itraconazol y fluconazol) y benzodiacepinas como el midazolam, alprazolam y el triazolam. Se ha reportado un aumento significativo e indeseable del efecto hipnótico (35). Con la benzodiacepina que no se reportan interacciones de este tipo es con el lorazepam, ya que esta no sufre metabolismo de fase I (citocromal) solo de fase II y no tiene metabolitos activos. Dentro del grupo de los macrólidos y azálidos, siempre se ha considerado a los de anillo de 14 átomos (eritromicina y claritromicina,) con mayor potencial para producir interacciones, que a los de 16 átomos (midecamicina, miocamicina, espiramicina) y al azálido azitromicina. Además, de inhibir el CYP3A4 la eritromicina y la claritromicina son inhibidores del CYP1A2 (24). Una excepción sería el compuesto de 14 átomos roxitromicina el cual tiene un bajo potencial de interacciones, aun así reduce el aclaramiento de la ciclosporina y teofilina (23). La diritromicina es otro macrólido con bajo potencial de interacciones, a pesar de esto hay que monitorear niveles de ciclosporina cuando se dan juntos (25).
La josamicina, macrólido
de 16 átomos, tiene la capacidad de interaccionar con teofilina,
sobre todo en niños, carbamazepina y ciclosporina (3).
La espiramicina no presenta prácticamente interacciones y puede
preferirse a la eritromicina en pacientes tomando ciclosporina. En el caso
de la miocamicina, esta si interacciona con ciclosporina y carbamazepina
(3). La eritromicina y la claritromicina interaccionan
con la teofilina, carbamazepina, ciclosporina, warfarina y su uso junto
con midazolam y triazolam debe ser evitado, si no es posible al menos reducir
la dosis de estos últimos en un 50% (27,29).
También no hay que olvidar que la patología que presente
un paciente puede influir en el grado de interacción de los medicamentos
con los citocromos. Se conoce que las infecciones virales y hacterianas
del tracto respiratorio muestran un efecto inhibidor sobre los citocromos,
especialmente el CYP1A2 (44). Se creé que es mediado por la acción
del interferón que se secreta durante estas infecciones (10).
Es también corriente usar macrólidos y azálidos para
el tratamiento de infecciones respiratorias por micoplasmas. En estos casos
el efecto inhibidor de los antibióticos sobre los citocromos podría
acentuarse a causa de la contribución del interferón (3).
Los inhibidores de la 3hidroxi-3-metilglutaril coenzyma A (HMG-CoA) reductasa
se utilizan para reducir los niveles altos de colesterol. La lovastatina
simprevastatina, y atorvastatina son metabolizados por el CYP 3A4, no así
la pravastatina y la fluvastatina. La fluvastatina es metabolizada por
el CYP2C9 y la pravastatina no es metabolizada por los citocromos. Las
estatinas pueden producir rabdomiolisis de una manera dosis dependiente
(35). Se han reportado interacciones de las estatinas
(rabdomiolisis) con itraconazol (200mg/día). Si se requiere
usar estatinas en un paciente bajo tratamiento con antifúngicos
azólicos, se debería preferir el uso de la pravastatina y
de la fluvastatina. Tanto la elaritromicina como la eritromicina, cuando
se utilizan juntamente con la lovastatina, se han asociado a rabdomiolisis
(43).
|
|
|
|
| CYP1A2 | ANTIPSICÓTICOS TÍPICOS | DISCINESIA TARDIA |
| CYP2C9 |
|
|
| CYP2C19 |
|
NEUROTOXICIDAD
PROLONGADA SEDACIÓN |
| CYP2D6 | ANTIARRÍTMICOS
BETA-BLOQUEADORES ANTIDEPRESIVOS TIRICÍCLICOS OPIODES FENFORMINA |
ARRITMIAS
BETA-BLOQUEADORES BRADICARDIA CONFUSIÓN
|
| CYP3A4 | DROGAS ANTILEUCEMIA | LEUCEMIA RELACIONADA
AL TRATAMIENTO |
INHIBIDORES SELECTIVOS DE LA RECAPTURA DE SEROTONINA (ISRS)
Los llamados inhibidores selectivos de la recaptura de serotonina son medicamentos de amplio uso en psiquiatría, no solo para el tratamiento de la depresión, sino en el tratamiento de diversos trastornos de ansiedad. Este tipo de sustancias pueden presentar interacciones de importancia clínica, pues se manifiestan como inhibidores de los citocromos. De los ISRS que presentan efecto sobre los citocromos se destacan: fluoxetina, paroxetina, sertralina, y la fluvoxamina. El citalopram, por otro lado, tiene un bajo perfil de interacciones y solo presenta un débil efecto (+) sobre el CYP2D6 (25). La fluoxetina tiene un fuerte efecto sobre el CYP 2D6 ( +++ ), menor sobre el CYP3A4 (++) y solo en dosis muy altas tiene Un efecto sobre el CYP1A2 (24,11). Este último no sería de importancia clínica. La fluvoxamina en cambio presenta un efecto moderado sobre el CYP3A4 y el CYP1A2 (++) y uno potente sobre el CYP2C9 (24,25). Además, es capaz de inhibir el CYP2D6 y el CYP2CI9(24). La sertralina presenta un débil efecto sobre el CYP2D6 y el CYP1A2 (+)y puede inhibir también el CYP2C9 y el CYP3A4.
La paroxetina es un fuerte
inhibidor del CYP 2D6 como la fluoxetina pero un débil inhibidor
del CYP3A4 (+) y en dosis altas puede inhibir el CYP1A2(24,25,11).
Ya esta última interacción sería de poca preocupación
en la clínica. No solo las drogas originales sino también
algunos metabolitos pueden comportarse como inhibidores enzimáticos.
La norfluoxetina, metabolito activo de la fluoxetina (vida media como de
10 días) es un inhibidor del CYP2D6(15) y del
CYP3A4(8). El haloperidol e un antipsicótico que
es metabolizado por varios citocromos, a saber: CYP1A2, CYP2D6 y el,CY
P3A4. El metabolismo por el CY P1A2 y el CYP2D6 es de menor importancia
que el CYP3A4. Cuando tenemos un medicamento de este tipo, pensamos que
las posibilidades de interacciones metabólicas se reducen mucho,
pues si se bloquea una enzima otras pueden seguir metabolizándolo.
Pero, que sucede cuando lo combinamos con un antidepresivo como la fluvoxamina.
Muy posiblemente una interacción, pues recordemos que la fluvoxamina
es un inhibidor del CYP1A2, CYP2D6 y del CYP3A4, es decir está inhibiendo
todas las vías metabólicas del haloperidol. La fluoxetina
por su parte también interacciona con el haloperidol aumentando
sus efectos extrapiramidales. En el caso del antipsicótico atípico
clozapina, este es metabolizado por el CYP1A2, el CYP3A4 y una menor vía
por el CYP2D6, de tal manera que es un fuerte candidato para interaccionar
con la fluvoxamina (8). La olanzapina es metabolizada
en parte por el CYP1A2 y puede interaccionar con la fluvoxamina también.
En el caso de que se manejen pacientes con depresión psicótica
y se emplee risperidona, hay que recordar que esta es degradada por el
CYP2D6 y su metabolismo es inhibido por la fluoxetina y la paroxetina (41).
Si la risperidona es la droga que se va a agregar hay que emplear una dosis
muy baja y aumentarla lentamente de acuerdo a la respuesta del paciente
(41). En este caso puede que se requieran dosis menores de la usual para
producir una respuesta. Además, la fluvoxamina interacciona con
la teofilina al bloquear su principal vía de metabolismo, el CYP1A2.
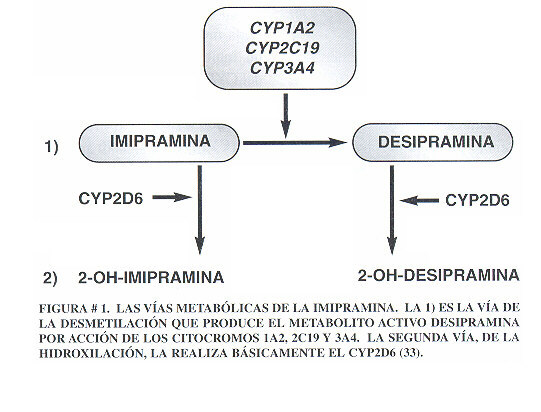
Como lo indican los estudios y se desprende de la figura
#1 (ver más adelante), el efecto de la fluvoxamina sobre el
metabolismo de la imipramina es más importante en la vía
de la desmetilación, donde inhibe todas las enzimas que participan
en ella. muestra menos efecto inhibidor sobre la vía de la hidroxilación
(8). Una interacción de características
similares a la explicada anteriormente se presentaría con la amitriptilina(38).
|
|
CYP2D6,CYP3A4 Y CYP2C9,2C19 CYP2D6 CYP2D6,CYP3A4 Y CYP2C9,2C19 CYP 1 A2,CYP3A4 Y CYP2C9,2C19 CYP3A4 |
| CELECOXIB
DICLOFENACO FLURBIPROFENO IBUPROFENO INDOMETACINA NAPROXENO PIROXICAM TENOXICAM |
CARVEDILOL
IRBESARTAN LOSARTAN GLIMEPIRIDA
FENITOÍNA |
FLUVASTATINA
MONTELUKAST METRONIDAZOL
|
ANTIEPILÉPTICOS
En esta sección estudiaremos los efectos que se producen cuando se mezclan entre si diferentes tipos de antiepilépticos. Los antiepilépticos, sobre todo los clásicos, son de los medicamentos que más interaccionan con los citocromos. El tipo de interacción con los citocromos se manifiesta de una manera dual en el caso de los antiepilépticos, pues algunos son inductores y otros inhibidores. Los antiepilépticos son inductores típicos y de los más importantes. Como ejemplos podemos mencionar: fenobarbital, fenitoína, primidona y carbamazepina. En el otro extremo se encuentra el ácido valproico, pues este medicamento se le considera un inhibidor enzimático. Además, el ácido valproico es capaz de inhibir la epóxido hidrolasa y la glucuronil transferasa (4). Precisamente de las acciones de los antiepilépticos sobre los citocromas derivan una serie de interacciones cuando estos, por necesidad, se combinan. Los antiepilépticos más recientes (vigabatrina, oxcarbazepina, topiramato, lamotrigina, gabapentina) fueron desarrollados para reducir al máximo posible las interacciones(40). Sin embargo, la oxcarbazepina, que es un pariente de la carbamazepina, y el topiramato producen cierto grado de interacción, pero aun así no alcanzan la magnitud de la interacción de los antiepilépticos Clásicos. Para que una interacción por inducción sea importante el medicamento que interacciona (el que se induce) debe tener un alto metabolismo, mayor del 75%, y un bajo índice terapéutico. La fenitoína es metabolizada por el CYP2C9 y por el CYP2C 19, de tal manera que inhibidores de estas enzimas interaccionan y pueden aumentar los niveles de fenitoína. En este caso hay que considerar la dosis del inhibidor que se está utilizando, su capacidad de inhibición y el tiempo y dosis de fenitoína, pues ella es un inductor del CYP2C9 y del CYP2C 19. El fluconazol, el ácido valproico, el ketoconazol (débil) y la amiodarona, son inhibidores del CYP2C9 y pueden aumentar los niveles de fenitoína (4).
También es posible que un antiepiléptico pueda interaccionar con otro por más de un mecanismo. En el caso de la fenitoína y el ácido valproico estos fármacos tienen un alto grado de unión a las proteínas y por lo general el ácido valproico puede desplazar a la fenitoína y reducir su concentración plasmática total, por lo menos al inicio del tratamiento. Esto puede producir un toxicidad transitoria, siempre y cuando los mecanismos de eliminación de la fenitoína no estén saturados, pues hay que recordar que la fenitoína es uno de los pocos medicamentos con una cinética saturable. Si aun no está en grado de saturación y a pesar de la reducción de la concentración total, la fracción de droga libre podría mantenerse y no hay necesidad de aumentar la dosis (9). Además, también hay que tomar en cuenta que la inhibición del valproato sobre el CYP2C9, que metaboliza la fenitoína, podría aumentar la concentración de ésta última. En el caso de estos dos fármacos es importante monitorear la fracción libre de fenitoína, que también puede calcularse de la concentración total mediante esta fórmula(40):
fenitoína libre =
[0.095 + 0.001 x Avc= concentración ácido valproíco
|
|
|
|
| ÁCIDO VALPROICO | CYP2C9, CYP2D6 |
|
| CARBAMAZEPINA | CYP1A2, CYP2C8,
CYP2C9, CYP2C19, CYP3A4 |
|
| ETOSUXIMIDA |
|
|
| FENITOÍNA | CYP1A2, CYP 2C8,
CYP2C9, CYP2CI9, CYP3A4 |
|
| FENOBARBITAL | CYP1A2, CYP 2CS,
CYP2C9, CYP2C 1 9 CYP3A4 |
|
| GABAPENTINA |
|
|
| LAMOTRIGINA |
|
|
| OXCARBAZEPINA | CYP3A4 |
|
| PRIMIDONA | CYP1A2, CYP2C8,
CYP2C9, CYP3A4 |
|
| TIAGABINA |
|
|
| TOPIRAMATO | CYP2C19 |
|
| VIGABATRINA |
|
|
En resumen la mezcla de estos
dos fármacos puede producir una disminución, aumento o ningún
cambio en la concentración total de la fenitoína (34).
Con lo que respecta al uso
concomitante de fenitoína y del fenobarbital, sabemos que los dos
fármacos son inductores enzimáticos y son metabolizados por
el CYP2C9 Y CYP2C19 (24). Por lo tanto al mezclarlos
se pueden inducir los dos y reducir sus niveles en sangre o puede haber
un aumento en los niveles de fenitoína o de fenobarbital. Esto último
sería el resultado de la acción sobre un mismo citocromo,
de tal manera que alguno de los dos podría actuar reduciendo el
metabolismo del otro por competencia. Claro, esto depende de las concentraciones
de ambos fármacos y de sus dosis. El topiramato es un antiepiléptico
capaz de inhibir el CYP2C19 lo que explica el aumento en los niveles de
fenitoína (25%) que se observan en algunos pacientes cuando estos
fármacos se mezclan (4). El topiramato no interaccionaría
con el fenobarbital pues el metabolismo de este a través del CYP2C19
es una vía menor. No se observan interacciones del topiramato con
otros antiepilépticos. Una de las pocas interacciones que presenta
la vigabatrina con los antiepilépticos es con respecto a la fenitoína.
La vigabatrina es capaz de reducir los niveles de fenitoína (25-40%)
en un tercio de los pacientes que consumen ambos medicamentos. Algunos
creen que la interacción es consecuencia del efecto inductor de
la vigabatrina sobre el CYP2C9 o el CYP2C19 (4).
La carbamazepina es un autoinductor de su propio metabolismo y en consecuencia la vida media de su uso crónico es mucho menor que la de su uso agudo(28). La carbamazepina, in vitro, es metabolizada por el CYP3A4 y en menor extensión por el CYP1A2 y el CYP2C8. La carbamazepina es extensamente metabolizada y menos del 1 % se excreta sin cambio en la orina. El principal metabolito es el 10- 11 -epóxido de carbamazepina y es activo. En monoterapia hace como el 25% de la dosis y en politerapia con otros antiepilépticos inductores hasta el 50% de la dosis (44). Cuando a la terapia con carbamazepina se hace necesario adicionar el ácido valproico, se observa un aumento en la toxicidad por carbamazepina, aun cuando los niveles de esta casi no aumenten (42). Lo que sucede es que el ácido valproico inhibe la epoxido hidrolasa, que es la enzima que metaboliza el 10- 11 epóxido de carbamazepina. En el caso del fenobarbital que tienen dos metabolitos importantes: el parahidroxifenobarbital (CYP2C9) y el N-glucósido de fenobarbital. La sola inhibición del CYP2C9 y la anulación de la producción del parahidroxifenobarbital, que es como de un 20%, no implicaría una interacción de importancia clínica. Esta interacción es importante cuando el fenobarbital se mezcla con ácido valproico, pues este también impide la formación del N-glucósido de fenobarbital (4). El ácido valproíco también interacciona con la lamotrigina pero no por una interacción con citocromos, sino a la manera como interacciona con el fenobarbital, es decir inhibiendo el metabolismo de fase dos tal como es la conjugación con glucurónido. En el caso de la lomotrigina esta es metabolizada en gran extensión por medio de glucuronidación(39). El uso conjunto de ácido valproico y topiramato parece que no presenta ninguna interacción de importancia clínica (39) a pesar de que el topiramato aumenta la beta oxidación del valproato(3). La carbamazepina, fenobarbital y fenitoína reducen los niveles de lamotrigina y topiramato (40-50%) cuando se mezclan. La oxcarbazepina también reduce los niveles de lamotrigina en sangre en cerca de un 29% (40).
Parte número dos de la segunda emisión. Esta es la segunda emisión de un trabajo científico sobre metabolismo de los medicamentos su importancia clínica. En esta segunda parte tratamos sobre las interacciones de interés más sobresalientes. Además de los mecanismos de interacción, nuestro objetivo es hacer conciencia, sobre la importancia del metabolismo de ciertos medicamentos.
BIBLIOGRAFIA
1- Li Albert P. y Malle Jurima-Romet. Overview: Pharmacokinetic Drug-Drug Interactions en: Advances in Pharmacology. Editores: August Thomas, Anders W., Murad Ferid y Coyle Joseph. Editorial Academic Press, San Diego. 1997;43:1-6.
2- Guengerich Peter. Role of cytochrome P450 Enzymes in Drug-Drug Interactions. En: Advances in Pharmakology. Editor: Thomas August et al. Academic Press, San Diego CA. 1997;43:7-36.
3- Anderson Gail. A mechanistic approach to antiepileptie drug interactions. Ann Pharmacother. 1998;32:554-63.
4- Hermann Bolt. Interactions between clinically used drugs and oral contraceptivas. Enviromental Health Perspectives. 1994;102 (supp19):35-38.
5- Back David y Orme L'E. Michael. Pharmacokinetic drug interactions with oral contraceptives. Clin Pharmacokinet. 1990; 18(6):472-484.
6- Michael Theodora. (2) Treatment-The establishe drugs. Pharmaceutical J. 1999;262:432-435.
7- Abernethy D.R. y Todd E. L. Impairment of caffeine clearance by chronic use of lowdose oestrogen-containing oral contraceptives. Eur J Clin Phartnacol. 1985;28:425-428.
8- Meyer Joette y Keith Rodvold. Drug interactions between nonsedating antihistamine and anti-infective agents. Infect Med 1996;13(7):609-613.
9- Piatek Ann y Lomaestro Ben. Update on drug interactions with azole antifungal agents Ann Pharmacother. 1998;32:915-928.
10- Gibaldi Milo. Drug interactions: Part I. Ann Pharmacother. 1992;May 26:709-713.
11- Nahata Milap y Abdel-Rahman. Oral terbinafine: a new antifungal agent. Ann Pharmacother. 1997;31:445-456.
12- Flórez J. Y Esplugues Y Farmacología de la motilidad del aparato digestivo. En: Farmacología humana de Flórez, 3 edición. Editorial Masson S.A, Barcelona, España. 1997. Pag. 736-738.
13- Kaumann Alberto. Do human atrial 5-HT4 receptors mediate arrhythmias? TiPS. 1994;dic. 15:451-455.
14- Granberry Mark y Gardner Stephanie. Erythromycin monotherapy associated with torsade de pointes. Ann Pharrnacother. 1996; jan 30:77-78.
15- Fichtl D. et al. Allgemaine Pharrnakologie und Toxikologie. Herasusgeber: Forth W., Henschler D., Rummel W. und Starke K. séptima edición. Spektrum. 1996. Pág. 52.
16- Flockhart, David. Drug interactions and the cytochrome P450 System. Clin. Pharmacokinet. 1995;29(suppl. 1): 45-52.
17- Pirmohamed Munir y Park Kevin. Genetic susceptibility to adverse drug reactions. Trends in Pharmacological Sciences. 2001;22(6):298-305.
18- Blum Robert et al. Effect of fluconazole on the disposition of phenytoin. Clin Pharmacol Ther. 1991;49:420-5.
19- Lacy Charles et al. Drug information handbook 2001-2002. Novena edición. Lexi-Comp Inc. Cleveland. Pag.1383.
20- Amsden Guy. Macrolides versus azalides: a Drug interaction update. Ann Pharmacother. 1995;29:906-917.
21- Luden Thomas. Pharmacokinetics interactions of the macrolide antibioties. Clinical Pharmacokinetics. 1985;10:63.79.
22- Jesen klosterskov et al. Possible interaction between oxcarbazepine and an oral contraceptive. Epilepsia. 1992;33(6):1149-1152.
23- Rosenfeld Willian et al. Effect of topiramate on the pharmacokinetics of an oral contraceptive containing norethindrone and ethinyl estradiol in patients with epilepsy. Epilepsia. 1997;38(3):317-323.
24- Kauffman Carol y Carver Peggy. Use of azoles for sustemic antifungal therapy. Editor: Thomas August et al. Advances in pharmacology. Academic Press. 1997 Vol 39. Pag 167-170.
25- Manjunath P Pai et al. Macrolide drug Interactions: an Update. Ann Pharmacother. 2000;34:495-513.
26-Chang K. C. et al. Altered theophylline pharmacokinetics during acute viral respiratory illness. Lancet. 1978; 1: 1132-3
27- Williams S. J. et al. Inhibition of theophylline metabolism by interferon. Lancet 1987;2:939-41.
28- Alderman Christopher y Allcroft Peter. Digoxin-ltraconazole interaction:Possible mechanisms. Ann Pharmacother. 1997;31:438-40.
29- The Medical Letter. Handbook of adverse drug interactions. 1990. pag 243.
30- Nemeroff, Charles et al. Newer antidepressants and the cytochrome P450 system. Am J Psychiatry. 1996; 153:311-320.
31- Laux G. et al. Pharrnakopsychiatrie tercera edición 2000. Urban&Fischer. Alernania. Pág. 193.
32- Gram Lars. Fluoxetine. New Engl J Med. 1994;17:1354-1361.
33- Brosen Kim. Drug interactions and the cytochrome P-450 system. Clin. Pharrnacokinet. 29(suppl. 1):20-25.1995.
34- Reisine Terry y Pastemak Gavril. En: Goodman & Gilman Las bases farmacológicas de la terapeútica. Novena edición Vol 1. Editorial Interamericana. 1996. Pág.572.
35- Granizo Enrique. Polimorfismo genético y sus aplicaciones en la farmacocinética de los opiodes. Vademécum farmacéutico 2000. Quinta edición. Edifarm Internacioiial. Pag.419-422.
36-Siegal Alan et al. Newer antidepressants and the cytochrome P450 system. Am J Psychiatry. 1996;153:311-320.
37- Ereshefsky Larry et al. Antidepressant drug Intractions and the citochrome P450 system. Clin Phartnacokinet. 1995;29 (suppl 1): 10-19.
38- Rosenhaum Jerrold. Managing selectiva serotonin reuptake inhibito-drug interactions in clinical practice. Clin Pharmacokinet. 1995; 29(suppl. 1):53-59.
39- Sabers Anne y gram Lennart. Newer anticonvulsansts: comparativa review of drug interactions and adverse effects. Drugs. 2000;60(1):23-33.
40- Brown
Adrian. Anticonvulsants: drug in-
* Dr. Ronald
González A. Ph.D. Depto. de Farmacología. Escuela de Medicina
Universidad de Costa Rica.