Myotonia Congenital: Clinic
characterization of a family affected by Thomsen's disease
Título corto: Miotonía
congénita en Costa Rica
Fernando Morales.1,2,
Patricia Cuenca1,2,
Gerardo del Vallé3,5,
Roberto Brian4, Mauricio
Sittenleld5,
Olga Montoya6,
Tetsuo Ashizawa7,
Alberto Rosa8 &
Keith Johnson9
Instituto de Investigaciones
en Salud1, Escuela
de Biologia2, Programa
de Investigación en Neurociencias, Universidad de Costa Rica; Neurolab3.
Curridabat; Servicio de Neurologia4,
Hospital Nacional de Niños; Servicio de Neurologia5,
Hospital San Juan de Dios; Clinica Oftalmológica6,
Curridabat; Departamento de Neurologia6,
Baylor College of Medicine, Houston, Texas, USA; Laboratorio de Neurogenética8,
Instituto de investigación Medica "Mercedes y Martin Ferereyra"
(INIMEC-CONICET), Córdoba, Argentina; Departamento de Genética
Molecular7, Anderson
College, Universidad de Glasgow, Escocia.
Congenital Myotonia is a muscular inherited diseases characterized by myotonia, hypertrophy, and stiffness. It 's inherited as autosomal dominant or recessive, known as Thomsen 's and Becker 's diseases,respectively. It is established the right clinical diagnosis of one family that previously was classified as myotonic dystrophy type 1. The clinical diagnosis was established after doing ocular,cardiac, physical and electromyographic tests. All of patients had normal strength but all of the patients showed myotonia, better showed in arms than hands, more evident in proximal muscles, and generalized myotonia that disappeared after repeating the movement. Clinical diagnosis must be followed by molecular diagnosis to confirm the clinical findings. These patients showed a clinical spectrum different from myotonic dystrophy, in harmony with a clinical diagnosis of Thomsen 's disease.
KEY WORDS: congenital myotonia, myotonic dystrophy, Thomsen 's disease, clinical management, genetic counseling.
RESUMEN
La miotonía congénita es una enfermedad muscular hereditaria caracterizada por excitabilidad incrementada de la fibra muscular, que se refleja en miotonía clínica, además de rigidez e hipertrofia. Su herencia puede ser de forma autosómica dominante o recesiva, designadas como miotonía generalizada de Thomsen y Becker respectivamente. Se establece el diagnóstico clínico correcto de una familia costarricense que previamente había sido diagnosticada con distrofia miotónica tipo 1, pero cuyo estudio a nivel molecular y clínico descartó una distrofia miotónica después de realizar exámenes físicos, electromiográficos, oculares y electrocardiograma a varios miembros de la familia. Los individuos poseían fuerza normal pero presentaban el fenómeno miotónico a nivel clínico y electromiográfico, el cual se apreciaba mejor en los brazos, era más evidente en la musculatura proximal que en la distal, además de ser generalizada e iba desapareciendo conforme el paciente repetía varia veces el movimiento. Ahora bien, con los estudios moleculares es necesario confirmar el diagnostico clínico propuesto en este trabajo. Los pacientes presentan un cuadro clínico claramente diferente de la DM1, compatible con miotonía congénita, y que por su forma de herencia; autosómica dominante; debe clasificarse como enfermedad de Thomsen.
ABREVIACIONES: ASMINE Asociación de pacientes Miasténicos y otras enfermedades - CLCNI Gen del canal de cloruro del músculo esquelético -CTG Citosina, timina, guanina-DM1 Distrofia Miotónica tipo 1 - DM2 Distrofia Miotónica tipo 2 - DMPK Proteína quinasa de la distrofia miotónica - MC Miotonía Congénita - PDM Distrofia Miotónica Proximal - PROMM Miopatía Miotónica Proximal
La palabra miotonía fue usada por primera vez en el año de 1891 y se refiere a un trastorno del músculo esquelético, que se caracteriza por una prolongación del tiempo de relajación de un músculo contraído voluntariamente o ante un estímulo mecánico, provocando una incapacidad transitoria para realizar el movimiento antagónico. Enfermedades asociadas con este trastorno son llamadas miotonías y de acuerdo con sus características clínicas se clasifican en: 1-canalopatías de sodio (paramiotonía congénita, parálisis periódica hipercalémica y miotonía potasio-agravado), 2-distrofias miotónicas (distrofia miotónica tipo 1 y 2 (DM1, DM2), miopatía miotónica proximal (PROMM), distrofia miotónica proximal (PDM) y 3- canalopatías de cloruro o miotonía congénita (enfermedad de Thomsen y enfermedad de Becker) (1,2,3). De estas condiciones miotónicas hay que mencionar que la más frecuente, la más estudiada y de la que se conoce más es la distrofia miotónica tipo 1; y dentro de las miotonías no distróficas, hay que mencionar que la más frecuente y la más estudiada es la miotonía congénita (4,5).
La miotonía congénita (MC) es una enfermedad muscular hereditaria, caracterizada electrofisiológicamente por presentar excitabilidad incrementada de la fibra muscular, que se refleja en miotonía clínica, además de rigidez e hipertrofia (6). El fenotipo clínico depende, en parte, si se hereda como una característica autosómica dominante o recesiva, designadas como miotonía generalizada de Thomsen y Becker respectivamente. Las dos enfermedades difieren clínicamente en la edad de manifestación, en una típica debilidad muscular transitoria vista solo en la forma recesiva, y en la amplitud de la miotonía (7). Los dos casos se asocian con mutaciones en el gen CLCN-1, ubicado en la región cromosómica 7q35 (8). Este gen está constituido por 23 exones, codifica para una proteína con doce dominios transmembrana que funcionan como un canal de cloruro en el músculo esquelético. Dicho canal es el principal responsable de la conductancia de la membrana en el músculo (9,10).
El inicio de las manifestaciones clínicas de la MC ocurre en la primera o segunda década de la vida. La miotonía es provocada por la contracción muscular, especialmente cuando se hace un esfuerzo físico repentino seguido de un periodo de descanso. La miotonía mejora con repetidas contracciones musculares y algunas veces es seguida por una debilidad que puede tardar hasta 30 minutos en la enfermedad de Becker (11). La rigidez muscular es temporal y puede afectar cada músculo esquelético del cuerpo. Típicamente los músculos miotónicos revelan un patrón característico en el electromiograma. La descarga miotónica se visualiza en el electromiógrafo (luego de una contracción voluntaria o de estimular el músculo eléctrica o mecánicamente) como una serie rítmica de potenciales que incrementan o decrecen progresivamente su amplitud, originando un ruido característico que recuerda los bombarderos de hélice utilizados en la segunda guerra mundial (12).
Ambas formas de la MC son causadas por mutaciones en el mismo gen. La búsqueda intensiva de mutaciones ha mostrado que la forma dominante es muy rara, ya que hasta la fecha menos de diez familias han sido caracterizadas a nivel molecular en el mundo. La forma recesiva es mucho más común, con una frecuencia entre 1/23.000 y 1/50.000. Los hombres aparentan estar afectados más frecuente-mente que las mujeres, en una proporción de 3: 1, cuando se toman en cuenta solamente los rasgos clínicos típicos. Sin embargo, los estudios familiares han revelado que las mujeres se ven afectadas en la misma frecuencia que los hombres, aunque con menor intensidad (5).
El objetivo de este trabajo fue caracterizar clínicamente una familia costarricense que previamente había sido diagnosticada como distrofia miotónica tipo 1, pero que resultó negativa para la mutación DMl (13).
Materiales y Métodos:
Pacientes
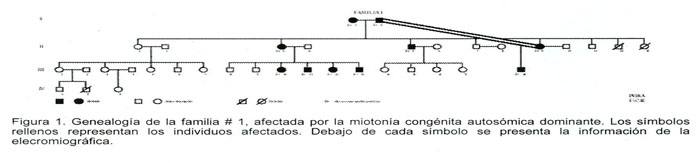
El estudio incluyó a 10 individuos de la familia designada como #1 (Figura 1), en la cual inicialmente se proponía el diagnóstico de una distrofia miotónica. La sospecha surgió por el nacimiento de un niño hipotónico sin causa aparente, con una madre que presentaba una electromiografia con descargas miotónicas típicas, sin embargo después de ser estudiados a nivel molecular, los pacientes resultaron negativos para la amplificación CTG en el gen DMPK de la DM1. Los individuos que inicialmente habían sido diagnosticados con DM1 eran el II.9 y el III.18. Todos los pacientes fueron traídos a la capital desde sus respectivos hogares debido a la escasez de recursos con los que cuentan y fueron hospedados en la Asociación de Pacientes Miasténicos y otras Enfermedades Neuromusculares (ASMINE) mientras se llevaban a cabo los estudios en la clínica Neurolab.
Diagnóstico clínico:
El diagnóstico clínico se estableció después de realizado los exámenes fisicos y electromiográficos. A los 10 pacientes se les realizó la electromiografia y solo a cuatro se les realizó un examen oftalmológico con lámpara de hendidura con el fin de buscar las cataratas características, en polvo de estrella, de los pacientes con DM1(4). Además, a todos los individuos se les practicó una evaluación electrocardiográfica.
Examen ocular mediante la prueba de la lámpara de hendidura (slit lamp test).
Con el fin de poder determinar la presencia y las características de las cataratas posibles en los individuos estudiados, se exploró la agudeza visual, la tensión ocular, el fondo del ojo, así como el segmento anterior en donde se determinó el reflejo pupilar y las características del cristalino. También se realizó un estudio fÍsico del ojo para determinar la presencia o ausencia de ptosis, la fuerza del músculo orbicular y los movimientos de los músculos extraoculares.
Examen clínico y electromiográfico
Se realizó una exploración neurológica completa con énfasis en los músculos, analizando la fuerza y la presencia del fenómeno miotónico ante la percusión muscular y en la fase de relajación después de una contracción voluntaria. Para evaluar la fuerza muscular se utilizó la escala MRC(14).
Además, se realizó un estudio electromiográfico convencional del músculo oponente del pulgar derecho en todos los pacientes y en algunos otros se exploraron músculos proximales de un miembro superior o inferior.
Resultados
El examen ocular realizado a los individuos I.1, I.2, II.9 Y III.18 dio como resultado la ausencia total de cataratas, de hecho, los resultados fueron completa-mente normales. Ninguno de los otros 8 individuos aquejó trastornos visuales, por lo que se decidió no hacerles la evaluación oftalmológica realizada a sus parientes.
Desde el punto de vista cardiovascular, no hay registros ni antecedentes familiares de problemas cardiovasculares, arritmias, palpitaciones, síncope, etc., y el electrocardiograma realizado a todos los 10 individuos dio como resultado un patrón normal.
Ninguno presentó dolores osteomusculares. El individuo I.2 aquejaba dolores en la espalda que no impresionó ser manifestación de la enfermedad, sino a estar relacionado a la edad (63 años) y las labores agrícolas que desempeña.
Solo un individuo (III.18)
tenía problemas en la marcha y no hubo quien presentara trastornos
sensitivos. El niño, quien resultó ser el más afectado
de todos los estudiados, presentó problemas motores después
de permanecer en reposo por un tiempo prolongado, sin embargo, conforme
se ejercitaba, estos problemas en la marcha iban desapareciendo poco a
poco. Los individuos II.9 y III.18 mostraron adiadococinesia. Estos mismos
individuos y el III.11 presentaron torpeza en las manos (se les dificultaba
maniobrar objetos con las manos, los cuales se les caían con frecuencia)
y el individuo III.18
relató que se caía
con frecuencia. El individuo II.5 presentaba calambres musculares.
Ninguno de los 10 individuos aquejó problemas digestivos crónicos ni del sueño, y solo tres de ellos presentaron apatía y retardo en el progreso escolar, sin embargo, no se hizo ningún examen al respecto.
El examen fisico logró determinar que todos los individuos presentaron fuerza normal, con la excepción del individuo II.18 quien presentó debilidad en la musculatura de cintura escapular y en los flexores del cuello.
Todos los individuos presentaron el fenómeno miotónico y el rodete miotónico con la excepción del individuo I.1. El fenómeno se apreciaba mejor en los brazos que en las manos y la miotonía era más evidente en la musculatura proximal que en la distal. La miotonía que se apreciaba en las manos era menos evidente y menos lenta de la que se observa en la DM1, aparte de que en éstos pacientes era generalizada y no en grupos de músculos. Los individuos II.9 y III.18 presentaron además miotonía ocular e hipertrofia de gemelos de pantorrilla. El paciente III.18 también presentaba el signo de Gowers, sin embargo mejoraba con las repeticiones del movimiento. Se encontraron reflejos miotáticos muy pobres en los individuos III.11 y III.18, y en el primero se encontraron además movimientos oculares fragmentados. La exploración de sensibilidad realizada a todos los individuos resultó ser normal en todas las modalidades. El paciente I.2 presentó asimetría de reflejos en los miembros inferiores atribuida a patología lumboradicular.
Con respecto al examen electromiográfico realizado a los 10 individuos de esta familia, hay que destacar que todos presentaron el patrón miotónico característico de una enfermedad miotónica.
Discusión
De acuerdo a los resultados obtenidos en este estudio, queda claro que el cuadro clínico de estos pacientes es claramente diferente de la DM1, siendo más compatible con miotonía congénita, y que por su forma de herencia; autosómica dominante; debe clasificarse como la Enfermedad de Thomsen. Cabe destacar, que este es el primer reporte clínico en Costa Rica de una familia afectada por la Enfermedad de Thomsen.
El diagnóstico clínico correcto de un trastorno neuromuscular hereditario como el tratado en este artículo, permite orientar los estudios moleculares que darán la confirmación definitiva del diagnóstico inicial, lo cual permitirá llevar a cabo un consejo genético y un manejo clínico adecuado de los pacientes.
En un reciente reporte (13), nosotros publicamos los primeros resultados moleculares sobre DM1 en Costa Rica. De las 21 familias analizadas, en 13 se pudo confirmar el diagnóstico clínico, mientras que en las otras 8 familias, el diagnóstico molecular resultó negativo, lo que indica que no padecen de DM1, por lo cual están siendo afectadas por otras condiciones miotónicas. Este es el reporte de la reevaluación clínica de una de esas 8 familias.
Estos pacientes presentaron
un cuadro clínico claramente diferente de la DM1, compatible con
el diagnóstico de miotonía congénita, que por su forma
de herencia autosómica dominante, debe clasificarse como la Enfermedad
de Thomsen. En el cuadro 1 se hace una comparación
de las características clínicas entre la distrofia miotónica
y la miotonía congénita. El cuadro 1
se elaboró con base en la información proveniente de diversas
fuentes, a saber Koty et al (2), Harper (4),
Lehmann-Horn & Jurkat-Rott (5), Zhang et al (6),
Meola (15), Walton (16) y a través
de la experiencia personal.
El diagnóstico clínico
correcto establecido para esta familia debe ser confirmado por métodos
moleculares.
En vista de que los individuos I.1 y I.2 resultaron clínicamente afectados, es de esperar que al menos dos diferentes mutaciones sean las que están segregando en ésta familia.
Se conocen varias mutaciones y su relación con el patrón de herencia de la MC. Algunas mutaciones se asocian con herencia autosómica recesiva y otras con una herencia autosómica dominante. Aunque algunas pueden comportarse como recesivas o dominantes, lo cual, probablemente se deba a la interacción de la mutación con el resto del genotipo de cada persona (7, 2). La lista de mutaciones ya descritas es de mucha utilidad como punto de partida para la caracterización molecular de las familias nuevas que van apareciendo.
El individuo III.18 resultó estar más severamente afectado que los demás miembros de la familia. Plassart-Schiess et al (17) estudiaron varias familias con MC y encontraron que en algunos casos, los individuos heterocigotas estaban menos afectados que los homocigotas para una determinada mutación, sugiriendo que el fenómeno de penetrancia incompleta y penetrancia reducida podría estar presente en algunas de estas familias. Por lo tanto, es probable que el individuo III.18 sea portador de dos mutaciones dominantes que le causan un cuadro clínico más severo y con manifestaciones desde el nacimiento.
Un adecuado diagnóstico clínico presuntivo permitiría decidir el tipo de mutaciones que se deben buscar en un primer esfuerzo del laboratorio de genética molecular. El diagnóstico clínico correcto obtenido en el menor tiempo posible y confirmado a través de los métodos moleculares es el mecanismo ideal para lograr un adecuado manejo clínico de los pacientes, lo que sin lugar a dudas contribuiría a lograr una mejor calidad de vida para ellos y sus familias. Los autores de este trabajo deseamos contribuir con el conocimiento clínico del diagnóstico diferencial del síndrome miotónico y hacemos un llamado a la necesidad de contar con estudios moleculares en el país para la confirmación diagnóstica de las enfermedades hereditarias.
Agradecimiento
Un agradecimiento muy especial
a ASMINE, en especial a la presidenta de su junta directiva Doña
Angelina Ramírez, por las facilidades brindadas para el alojamiento
de esta familia en San José. A Fernando Ortíz, Melissa Vásquez,
Warner Alpízar y María Auxiliadora González por su
colaboración en la atención de la familia en Sán José.
El Prof. Johnson visitó Costa Rica gracias al apoyo brindado por
el Programa de Académicos y Visitantes de la Oficina de Asuntos
Internacionsles de la Universidad de Costa Rica. La Wellcome Trust colaboró
con el mantenimiento de la familia en San José.
Referencias
1. Meola G. Myotonic dystrophies. Curr. Opin. Neurol. 2000 13: 519-525.
2. Koty P., Pegoraro E., Hobson G., Marks H.G., Turel A., Flagler D., Cadaldini M., Angelini C., Hoffman E. Myotonia and the muscle chloride channel: dominant mutations show variable penetrance andfounder effect. Neurology. 1996 47: 963-968.
3. Torres L., Velez M., Cosentino C. Becker's myotonia in Peru. Rev. Neurol. 2000 30:1033-1036.
4. Harper PS. "Myotonic dustrophy". 1989. 2nd ed. Saunders, Philadelphia, USA.
5. Lehmann-Horn F. & Jurkat-Rott K. Voltage-gated ion channel and hereditary disease. Physiol. Rev. 1999 79: 1317-1372.
6. Zhang J., George, Jr A., Griggs R., Fouad G., Roberts J., Kwiecinski H., Connolly A., Ptácek. Mutations in the human skeletal muscle chloride channel gene (CLCN1) associated with dominant and recessive myotonia congenita. Neurology. 1996 47: 993-998.
7. Meyer-Kleine C., Steimeyer K., Ricker K., Jentsh T., Koch M. The spectrum in the mayor human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am. J. Hum. Genet. 1995 57: 1325-1334.
8. Koch M., Steimeyer K., Lorenz C., Ricker K., Wolf F., Otto M., Zoll B., Lehmann-Horn F., Grzeschik K., Jentsch T., The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992 257: 797-800.
9. Esteban J., Neumeyer A., Mckenna-Yasek D., Brown R. Identification of two mutations and a polymorphism in the chloride channel CLCN-1 in patients with Becker's generalized myotonia. Neurogenetics. 19981: 185-188.
10. Zhang J., Sanguinetti M., Kwiecinski H., Ptacek L. Mechanism of inverted activation of CLC-1 channel caused by a novel myotonia congenita mutation. J. Biol. Chem. 2000 275: 2999-3005.
11. Tawil R., Griggs R., Rose M. Chabbelopathies. In: Pulst S., ed. Neurogenetics. New York, USA. Oxford University Press. 2000. p. 51-52.
12. Kimura, Jun. 2001. Electrodiagnosis in disease of nerve and muscle: principles and practice. 3rd Edition. Oxford University Press, Inc. New York, USA; pp: 343.
13. Morales F., Cuenca P., Brian R., Sittenfeld M. & del Valle G.Diagnóstico molecular de la Distrofia Miotónica (DM) en Costa Rica. ACM 2001; 43: 159-167.
14. Medical Research Council. 1976. Aids to the examination of the peripheral nervous system. Memorandum no. 45 (H.M.S.O: London).
15. Meola G. Clinical and genetic heterogeneity in myotonic dystrophies. Muscle & Nerve. 2000 23: 1789-1799.
16. Walton, John. 1981. Disorders of voluntary muscle. 4th Edition. Churchill Livingstone Inc. New York, USA; pp: 508-513.
17. Plassart-Sehiess
M., Gervais A., Eymard B., Lagueny A., Pouget J., Warter J., Fardeau M.,
Jentsch T. & Fontaine B. Novel muscle chloride channel (CLCN1) mutations
in myotonia congenita with various modes of inheritance including incomplete
dominance and penetrance. Neurology. 1998 50: 1176-1179.
Cuadro 1: Similitudes y diferencias entre la distrofia y la miotonía congénita
| STEINER |
|
|||
|
|
|
|
de Thomsen |
de Becker
|
| Atrofia muscular |
|
|
|
|
| Hipertrofia |
|
|
|
|
| Rigidez |
|
|
|
|
| Debilidad muscular |
|
|
|
|
| Debilidad de la musculatura facial |
|
|
|
|
| Debilidad de los músculos mandibulares |
|
|
|
|
| Debilidad de la musculatura proximal de las piernas |
|
|
|
|
| Debilidad de la musculatura distal de miembros |
|
|
|
|
| Degeneración muscular |
|
|
|
|
| Miotonia (EMG) |
|
|
|
|
| Miotonía |
|
|
|
|
| Problemas cardiacos |
|
|
|
|
| Progresión de la enfermedad |
|
|
|
|
| Ptosis |
|
|
|
|
| Cataratas |
|
|
|
|
| Calvicie frontal |
|
|
|
|
| Cara en forma de hacha |
|
|
|
|
| Retardo mental |
|
|
|
|
| Otros sistemas |
|
|
|
|
| Dolor muscular |
|
|
|
|
| Edad de manifestación |
|
|
|
|
| Forma congénita |
|
|
|
|
| Forma de herencia |
|
|
|
|
|
|
|
|
|
|
| Anticipación |
|
|
|
|
+ hallazgo presente; ++ hallazgo presente y muy propminente; (+) hallazgo presente pero enmenor grado;-hallazgo no presente; ? no conocido