SUMMARY
Immunologic marker studies of the lymphoid leukemias have greatly improved the precision of diagnostics of the disorders by providing specific information regarding the lineage and stage of maturation of the malignant cells. Such studies have also enhanced our understanding of normal lymphocite development, permitting reproducible identification of lymphoid cells in discrete developmental stages. The LLA is an immunophenotypically heterogeneous disease with clinically important subtypes representing clonal expansions of lymphoblasts at different stages of maturation. Knowledge of immunologic features of leukemic cells has been essential for the generation of phenotype - specific response data in the context of modern therapy for LLA. In the chronic lymphoproliferative disorders, although the information derived from immunophetyping is essential to define the type of the lymphoid population and is hepful to distinguish between the varios conditions, the clinical, cytological and histopathological features of the disease are also important in order to establish a more precise diagnostic.
INTRODUCCION
El empleo de la citometría de flujo en el estudio de los Síndromes Linfoproliferativos crónicos (SLC) posee especial relevancia, al permitir un diagnóstico diferencial rápido entre una linfocitosis reactiva y un proceso monoclonal y en este caso contribuir a su filiación diagnóstico. El inmunofenotipo permite la clasificación diagnóstica de los SLC en neoplasias de origen B, T y NK (20). Además dentro de cada uno de estos subtipos de hemopatías se han podido definir subgrupos fenotípicamente diferentes con características clínico-biológicas específicas. El análisis de marcadores inmunológicos de leucemias y de Linfomsa no Hodgkin (LNH) permite una clasificación reproducible de esas malignidades. Así, la caracterización de estas malignidades se hace de acuerdo a criterios clínicos, morfológicos e inmunológicos.
DESORDENES LINFOPROLIFERATIVOS B
Dos grupos principales son considerados: las leucemias propiamente y la fase leucémica del LNH. La clasificación es la siguiente: (15)
LEUCEMIAS:
Leucemia linfocítica crónica
B
a) tipo común (LLC-B)
b) con > 10% de prolinfocitos (LLCIPL)
Leucemia prolinfocítica B (LP-B)
( > 55% de prolinfocitos)
Leucemia de células peludas
a) forma clásica ( HCL)
b) variante ( HLC-V)
Leucemia de células plasmáticas
( PCL)
a) linfoplasmacitica
b) forma diferenciada
SINDROMES LINFOMA/LEUCEMIA:
A) LEUCEMIA LINFOCITICA CRONICA B:
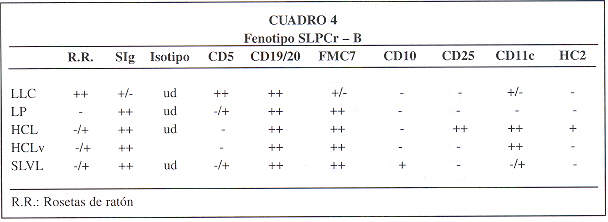
Se caracteriza por mostrar una infiltración periférico masiva por células que expresan los marcadores B: slg ( inmunoglobulina de superficie), CD19, CD20 y CD22, con intensidad inferior a los linfocitos B maduros normales de sangre periférico; son habitualmente CD5 + y CD23 + y carecen de reactividad para los antígenos FMC7, CD 1 0 y CD 103. La leucemia linfocítica crónica con prolinfocitos y la forma variante de tricoleucemia tienen características fenotípicas intermedias entre la leucemia linfocítica crónica (LLC) y la leucemia prolinfocítica (LPL) y entre la leucemia prolinfocítica y la tricoleucemia clásica, respectivamente (20).
B) LEUCEMIA PROLINFOCITICA B :
El prolinfocito B, aunque de apariencia morfológica más inmadura que el linfocito de la LLC, presenta características antigénicas de mayor grado de diferenciación: expresión de antígenos FMC7 y CD22, slg de alta densidad y menor proporción de rosetas de ratón y de antígeno CD5 (8,18,25), siendo por otro lado positivo para CDlc (4).
C) LEUCEMIA DE CELULAS PELUDAS (HCL):
Inicialmente el origen de las células
peludas o tricoleucocito fue muy debatido sugiriéndose incluso una
ontogenia monocítica debido a su capacidad fagocítica y adherencia
al plástico en cultivos; sin embargo, hoy es incuestionable su origen
linfoide B basado en la expresión de slg, antígenos B (CD19,
CD20, CD22) y reordenamientos de los genes de las inmunoglobulinas. Los
estadíos inmunofenotípicos revelan un estado madurativo avanzado,
posterior al de la LLC-B y próximo al de la LP-B, dado el isotípo
de las slg y alta expresión de FCM7, junto a la baja capacidad para
formar rosetas con hematíes de ratón y de coexpresar el antígeno
CD5 (16). La existencia de fenotipos T en la tricoleucemia
es cuestionable ya que los pocos casos descritos son previos a la disponibilidad
de AcMc. No obstante, recientemente se han comunicado dos casos T asociados
a infección por HTLV II (19).
C) LEUCEMIA DE CELULAS PLASMATICAS (PCL):
Es una rara pero seria enfermedad cuya posición nosológica es difícil en cualquier clasificación. El criterio morfológico e inmunológico indican que las células están en el estado final de la maduración de las células B. Se debe tener cuidado en interpretar los resultados de los estudios de marcadores ya que estas células plasmáticas como su contraparte normal, carecen de los antígenos de clase II de histocompatibilidad (la) en la membrana, de la mayoría de los, antígenos de células B (CD19, CD20, CD21) y también son negativos para Smlg. La combinación típica de marcadores positivos en la leucemia de células plasmáticas (LCP) es: CD38 (OKT10) +, Cyt Ig + (Kappa o Lamda) y reactividad con alguno de los antígenos asociados a las células plasmáticas: PCA-1, BU 11, Ri3, etc (14). Algunos de los casos con células linfoplasmaticas pueden todavía retener algunos de los marcadores B tardíos, como FMC7 o Smlg, pero son siempre CD38 +, el principal antígeno de las células plasmáticas. Una rara forma de PCL semeja una leucemia aguda; las células son blastos y pueden tener o no algunas características morfológicas ( por microscopía de luz) de plasmablastos. Su fenotipo es positivo solo para CD38 (OKT10) y Cyt I g. La microscopía electrónica puede ser esencial para aclarar el diagnóstico en algunos de estos casos, particularmente aquellos con características linfoplasmacitarias o plasmablásticas (14).
D) SINDROMES LINFOMALEUCEMIA DE CELULAS B:
En los LNH la presencia de linfocitos T y B normales en las adenopatías tumorales es un obstáculo para la identificación de las células neoplásicas. Sin embargo, el análisis simultáneo de varios marcadores y la cuantificación de ADN, dada la elevada incidencia de aneuploidías es esta enfermedad, permite obviar esta dificultad. Así, ha podido demostrarse que los LNH-B de linfocitos pequeños ( linfoma linfocftico bien diferenciado difuso de bajo grado de malignidad) coexpresan los antígenos CD19, CD20, HLA/DR, slg (tenue, de baja densidad), CD5 y CD21. Los linfomas centrocítico y centrocítico/centroblástico de pronóstico intermedio, presentan menor positividad para el antígeno CD5, suelen ser FMC7 +, CD 1 0 + /- y expresan sIg abundantemente (22). Los linfomas centroblástico e inmunoblástico, de mediano y alto grado de malignidad, respectivamente, se dividen en cuatro grupos fenotípicos cuya relevancia clínica está aún por demostrarse: (22)
1) CD19 +, CD20 +, slg + CD21-
2) CD 19 +, CD20 +, slg CD21 +
3) CD19 +, CD20 +, slg + CD21-
4) CD20 +, slg +, CD19 -, CD21-
FASE LEUCEMICA DEL LINFOMA FOLICULAR (FL):
El FL es el tipo más común de LNH. La enfermedad se origina en el centro germinal de los ganglios linfáticos y frecuentemente la médula ósea esta involucrada en algún grado, lo cual puede ser mejor reconocido por medio de biopsia. Este linfoma puede estar involucrado o aún presentarse en fase leucémica en una proporción de pacientes, quienes pueden ser erróneamente diagnosticados como LLC especialmente cuando el cómputo de leucocitos es muy alto. El inmunofenotípo es importante para establecer la clonalidad de la linfocitosis periférico y para ayudar a distinguir las células FL de aquellas de otras leucemias de células B (18). A diferencia de las células de la LLC, los linfocitos FL expresan fuertemente la Smlg monoclonal y son reactivas con el AcMc CD10 y FMC7 y usualmente no reactivas con CD5. La histología del ganglio linfático es esencial para establecer el diagnóstico de FL y la biopsia de la médula ósea puede ser útil para mostrar una infiltración linfoide paratrabecular (14,15).
LINFOMA ESPLENICO VELLOSO (SLVL):
Es una enfermedad caracterizada por gran esplenomegalia sin adenopatías o sólo localizadas (25% de los casos) y linfocitos con prolongaciones citoplasmáticas, que se presenta en personas de más de 55 años de edad (edad media 70 años) (2,28). En la mayoría de los casos hay una leucocitosis moderada (10 a 30 x 109 / L) con una linfocitosis que se acompaña de anemia y trombocitopenia leve en la mayoría de los enfermos. Un dato característico es la presencia de un componente monoclonal sérico (dos tercios de los SLVL), generalmente IgM, acompañado a veces de cadenas ligeras en la orina (14).
Las características claves para su diagnóstico son:
1) La caracterización en sangre periférico de células linfoides con la morfología característica y un fenotipo B con expresión intensa de slg, CD19, CD20, CD22, CD24. La presencia del antígeno FMC7 junto a la frecuente ausencia del antígeno CD5 indica un estadío posterior a la LLC. El SLVL suele ser CD 10 + y a diferencia de la leucermia de células peludas es negativo para CD25, B-Iy7, HC-2 y generalmente CD11c (14,15)
2) Hallazgos citológicos e histológicos en médula ósea. A diferencia de la HCL, la médula ósea es fácilmente aspirada, de un moderada a un bajo grado de infiltración linfoide, frecuentemente con un patrón nodular.
3) La histología del bazo demuestra una infiltración de la pulpa blanca y un menor grado de infiltración de la pulpa roja, diferente a la HCL o sus variantes en donde solo la pulpa roja esta involucrada.
FASE LEUCEMICA DEL LNH DE LA ZONA DEL MANTO / INTERMEDIO (I-LNH):
Este linfoma se origina a partir de la zona del manto de los folículos de los ganglios linfáticos y esta caracterizado por un patrón de infiltración predominantemente difuso y/o vagamente nodular. En la Formulación de Trabajo para la clasificación de LNH, el I-LNH puede ser asignado a la categoría de tipo hendido pequeño difuso con un pronóstico intermedio. Esta enfermedad se presenta principalmente en personas añosas, con linfadenopatía y esplenomegalia. La sangre periférica no es frecuentemente involucrada. El cuadro morfológico sanguíneo es pleomófico con un espectro de tamaños celulares y configuración nuclear. Los marcadores de membrana muestran que las células reaccionan en la mayoría de los casos con los marcadores pan-B y son FMC-7 +. Como en la LLC, la expresión del antígeno p67 reconocido por el AcMc CD5 es una característica consistente: algunas veces las células forman rosetas de ratón y una proporción variable de casos son CDI10+. De acuerdo a las observaciones de varios autores, ningún caso de LNH-I llena el perfil inmunológico característico de LLC (15). La histología del bazo y el ganglio linfático del LNH-I muestran un patrón de infiltración difusa por células linfoides pleomórficas con centros germinales residuales desnudos. La biopsia de médula ósea demuestra en la fase leucémica una infiltración fuerte difusa por linfocitos de tamaño pequeño a mediano con la presencia de histiocitos epitelioides rosados.
FASE LEUCEMICA DE LNH-B DE CELULAS GRANDES:
Este corresponde al LNH centroblástico o inrnunoblástico. La sangre periférica está infrecuentemente involucrada en éste LNH. Los marcadores de membrana son esenciales para diferenciar los LNH de células grandes de naturaleza B o T y en algunos casos es necesario confirmar por medio de análisis moleculares de los genes de las cadenas de las inmunoglobulinas o el TCR (15). Una minoría de LNH Ki-1 + corresponde a éste grupo de LNH-B de células grandes.
DESORDENES LINFOPROLI-FERATIVOS T
Estos constituyen un espectro de enfermedades caracterizados por la proliferación clonal de linfocitos T maduros (post-tímicos). La clasificación de estos desórdenes es la siguiente ( 14,15):
A) LEUCEMIA LINFOCITICA CRONICA T:
Es descrita como madura debido a que su fenotipo de membrana corresponde al de los linfocitos posttímicos ( TdT y CD I son siempre negativos en estas células). Los únicos dos marcadores pan-T consistentemente positivos son CD2 (o rosetas E) y OKT17 ( este anticuerpo monoclonal no es frecuentemente usado). Otros como CD3 o CD5 ( negativo en LLC-T) o CD7 (frecuentemente negativas en Leucemia/ Linfoma de células T adultas y Síndrome de Sesary) no reaccionan como es lo esperado en linfocitos T maduros normales (sangre periférico). La mayoría de los casos de LLC-T ( o LGL- leucemia) son proliferaciones CD8 +, Leu 7 +, mientras que las otras malignidades son CD4+ principalmente (14).
B) LEUCEMIA PROLINFOCITICA T (LP-T):
Los prolinfocitos T exhiben un fenotipo post-tímico ( CD 1 - , TdT ) con expresión de los marcadores pan-T: CD2 +, CD3 +, CD5 +, CD7 + . Debe destacarse que éste último antígeno (CD7) suele ser negativo o de baja expresión en los otros SLP-T maduros. Aunque en la mayoría de los casos (70 %) el fenotipo de la LP-T es CD4 +, CD8 - , hay también casos CD4 +, CD8 + (20 %) y CD4 - CD8 + (10%) (8,24).
C) LEUCEMIA/ LINFOMA DE CELULAS T ADULTAS (LLTA):
La LLAT presenta un fenotipo T maduro, en la que destaca la frecuente ausencia de dos marcadores pan-T: CD7 (negativo en el 90% de los casos) y el CD3 (expresión variable), sin embargo, éstas células son positivas para el CD2 y el CD5. Generalmente tienen fenotipo CD4 +, CD8 -, siendo típica, aunque no exclusiva, la alta expresión de CD25 (receptor de la interleucina 2) (8, 23, 27), habiéndose postulado que éste receptor pueda contribuir a la expansión del clon tumoral, en relación con el HTLV-I.

C) LEUCEMIAS DE CELULAS GRANULARES:
Las proliferaciones de linfocitos grandes granulares (LGG) constituyen un área de notable controversia como lo demuestran las múltiples denominaciones que han recibido: linfocitosis crónica T, linfocitosis granular crónica, LLC-T de células supresoras, enfermedad T-gamma (28-29). Aunque los términos proliferación de LGG y de células NK se han considerado sinónimos, probablemente las leucemias de células NK representan un subgrupo minoritario (10-20%) dentro del conjunto de las leucemias de LGG, como se deduce de las diferencias fenotípicas y moleculares. El 88% de las proliferaciones de LGG son CD2 +,CD3 + mientras que el 12% restante son CD2 + CD3 -. Las primeras por tanto corresponderían a leucemias T post- tímicas, mientras que las segundas serían genuinas leucemias de células NK. Los autores Richard & Scott descubren que dentro del grupo mayoritario (CD3 +) el 80% son CD8 + y el 20% restantes se distribuyen en partes iguales entre casos CD4 + CD8 - , CD4 + CD8 + y CD4 - CD8 -. En el 60-80% de estos enfermos los LGG son CD16 + y CD57 + pero al parecer, menos del 20% expresan CD56. Por el contrario, las leucemias de células NK (CD3-) se caracterizan por expresar los antígenos asociados a actividad NK ( CD56 y CD16). Así mismo, se han descrito casos excepcionales de LLA (TdT+) con fenotipo NK (28 -29).
D) MICOSIS FUNGOIDE (MF) Y SINDROME DE SESARY (SS):
El SS constituye, junto con la MF, la presentación genuina de los linfomas cutáneos, siendo considerado por muchos autores como la variante leucémica de la MF. Ambos son linfomas T post-tímicos. Clásicamente, los pacientes con MF, tras una fase inicial premicótica, permanecen en fase cutánea durante años o incluso décadas. La progresión se hace evidente cuando aparecen tumores cutáneos ulcerados, con diseminación a ganglios linfáticos superficiales y otros órganos internos (fase tumoral). Solo un 10% de los pacientes con MF se leucemizan, pasando a ser entonces un SS. En casos excepcionales los enfermos con MF desarrollan la fase tumoral sin pasar por la fase previa de placas cutáneas (27-28). El fenotipo es T maduro CD 1, - CD2 + CD3 + , CD5 + generalmente con expresión exclusiva de CD4, aunque se ha descrito algún caso CD4 + CD8 +. A diferencia de la LP-T y la LLTA suele ser negativo para CD7 y CD25, respectivamente (2, 26). El grupo de Catovsky ha señalado la existencia de casos excepcionales de leucemia con morfología de SS pero sen infiltración cutánea y fenotipo distinto, ya que suelen ser CD4 + CD8 + o CD4 - CD8 +, por lo que probablemente se trata de una entidad diferente.
E) LINFOMAS NO HODGKIN DE CELULAS T:
Clínicamente son muy agresivos. Las características clínicas son hepatoesplenomegalia, linfadenopatía y frecuentemente infiltración de tejidos no linfoides. La piel, la sangre periférico y la médula ósea pueden estar involucrados de una forma secundaria. Histológicamente los LNH-T corresponden a un tipo difuso de LNH, aunque puede haber una infiltración preferencial o aún exclusiva de la zona paracortical de los ganglios linfáticos. Morfológica y fenotípicamente los LNH-T son muy heterogéneos, pueden presentarse células grandes con características de inmunoblastos La designación de LNH-T incluye un número de entidades tales como la LLTA, el linfoma de Lennert con su patrón histopatológico distinto y casos de linfadenopatía angioinmunoblástica, como es demostrado por el análisis de ADN de los genes de la cadena TCR-b. Los marcadores de membrana demuestran que las células del LNH-T tienen un fenotipo de célula T madura ( TdT- ,CD1a - ) y corresponde a proliferaciones CD4 + o CD8+ o puede reaccionar con estos dos AcMc.
Las células con características inmunoblásticas son frecuentemente no reactivas con uno o más marcadores pan-T ( CD5 O CD7) y pueden expresar antígenos de activación de células T reconocidos por los AcMc CD38, CD25, Ki- 1 y los antígenos de histocompatibilidad de clase II. Es de interés la carencia de clonalidad por el análisis del ADN en algunos de estos casos, a pesar del curso clínico agresivo y de la presencia de anormalidades cromosómicas (26). Se ha sugerido la existencia de cuatro grupos de acuerdo con la expresión de los antígenos CD4 y CD8 (22):
1) inmaduro ( CD4 -, CD8 - )
2) cooperador ( CD4 +)
3) supresor (CD8 + )
4) indefinido (CD4 +, CD8 +)
De ellos el grupo CD4 + sería el más frecuente y con mejor evolución clínica.
RESUMEN
Los estudios de los marcadores de las leucemias linfoides han mejorado enormemente la precisión del diagnóstico, ya que proveen información específica que toma en cuenta el linaje y el estado de maduración de las células malignas. Tales estudios han aumentado nuestro conocimiento del desarrollo normal de los linfocitos, permitiendo la identificación reproducible de las células linfoides en los estados de desarrollo. La LLA es una enfermedad heterogénea con subtipos clínicamente importantes representando expansiones clonales de linfoblastos en los diferentes estados de maduración. El conocimiento de las características inmunológicas de las células leucémicas ha sido esencial para la generación de la respuesta fenotipo específica en el contexto de la terapia moderna para la LLA. El los desórdenes linfoproliferativos crónicos, aunque la información derivada del inmunofenotipo es esencial para definir el tipo de población linfoide y es útil para distinguir entre varias poblaciones, la clínica, citología y las características histopatológicas de las enfermedad también son importantes para establecer un diagnóstico más preciso.
BIBLIOGRAFIA
1) Alfsen GC, Beiske K, Holte H, Hovig E, Deggerdal A, Sandlie I, Widing E, Slordahi S, Klepper IK, Sizoo W, Van Der Elsen P, Bolhuis RLH. T- cell receptor g d/ CD3+ 4 - 8- T cell acute lymphoblastie leukemias: a distinct subgroup of leukemias in children. A report of five cases. Blood 1991. 77 : 2023-2030.
2) Bennett JM, Catovsky D, Daniel MT, Faldrin G, Galton DAG, Gralnick HR, Sultan C. Proposals for the classification of chronic (mature) B and T lymphoid leukemias. Journal Clinical Pathol 1989, 42 : 567-584.
3) Brenner MK, Hoffbrand AV. 1990. Normal lymphocytes and their bening disorder. pp : 323-338. In Hoffbrand & Lewis. 3a edición. Postgraduate Haematology. Heinemann Medical Books. London.
4) Catovsky D. 1990. Chronic lymphoid leukemia, pp:418-450. In Hoffbrand & Lewis. 3a ed. Postgraduate Haematology. Heinemann Medical Books. London.
5) Dexter HG, Thiel E, Ludwig W-D. Review of the incidence and clinical relevance of myeloid antigen positive in acute lymphoblastic leukemia. Leukemia , 1991, 5:637-645.
6) Garant R, Béné M, GEIL. A new approach of acute lymphoblastic leukemia immunophenotypic classification : 1984-1994. The GEIL experience. Leukemia and lymphoma 1994 13 :1-5.
7) Janossy B, Coustan-Smith E, Camparia D. The reliability of cytoplasmic CD3 and CD22 antigen expression in the immunodiagnosis of acute leukemia,: a study of 500 cases. Leukemia 1989, 3: 170- 181.
8) Jones RA, Master PS, Child JA, Roberts BE, Scott CS. Diagnostic differentiation of chronic B-cell malignancies using monoclonal antibody L161 (CD1c). British Journal of Haematology, 1989, 71 : 43-46.
9) Lauria F, Raspadori D, Martinelli G, el al. Increased expression of mieloid antigen markers in adult acute lymphoblastic leukemia patients : diagnostic and prognostic implications. British Journal of Haematology, 1994, 87 : 286-292.
10) López de Macedo A. 1994. Estudio antigénico de la hematopoyesis normal y sus implicaciones en la detección de enfermedad mínima residual en leucemias agudas mieloblásticas. Departamento de Medicina, Universidad de Salamanca. pp : 16-20.
11) Ludwig WD, Harbott J, Batram CR, Komischke B, Sperling C, Teichmann JV, SeibtJung H, Notter M, Odenwald E, Nehmer A, Thiel E, Riehm H. 1993. Incidence and prognostic significance of immunofenptypic subgroups in childhood acute lymphoblastic leukemia : Experience of the BFM study 86. 131:269-282 In Recent results in cancer research. Edited by W.D. Ludwig and E.Thiel. Springer-Verlag, Berlin, Heidelberg.
12) Macintyre EA, Salloum E, Sigaux F. Comparison of a lb and g /d expressing CD3+ acute lymphoblastie leukemias. Nouvelle Revue Francaise d'Hematologie 1990, 32 :95-99.
13) Matutes E, Worner I, Sainati L, De Oliveira MP, Catovsky D. Advances in the lymphoproliferative disorders. Review of our experience in the study over 1000 cases. Biology Clinical Hematology, 1989 ; 11 : 53-62.
14) Matutes E, Brito-Bapapule V, Swansbury J, Ellis J, Morilla R, Dearden C, Sempere A, Catovsky D. Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood 1991, 78 : 33269-3274.
15) Melo JV, Catovsky D, Galton DAG. The relationship between chronic lymphocytic leukemia and prolymphocytic leukemia. I. Clinical and laboratory features of 300 patients and characterization of an intermediate group. British Journal of Haematology, 1986, 63 : 377-387.
16) Melo JV, Megde U, Parreira A, Thompson I , Lampert IA, Catovsky D. Splenic B cell Lymphoma with circulating villous lymphocytes : differential diagnosis of B cell leukemias with large spleens. Journal Clinical Pathology. 1987, 40 : 642-651.
17) Melo JV, Parreira A. 1992. Leucemia prolinfocítica B. pp : 323-329. En López Borrasca et al. Enciclopedia de Hematología Iberoamericano. Universidad de Salamanca.
18) Melo JV, San Miguel JF, Moss VE, Catovsky D. The membrane phenotype of hair cells : A study with monoclonal antibodies. Semm. Oncol, 1984,11 : 381-385.
19) Mulligan SP, Matutes E, Dearden C, Catovsky D. Splenic lymphoma with villous lymphocytes : natural history and response to therapy in 50 cases. British Journal of Haematology 1991, 78 : 206-209.
20) Orfao A, Gonzáles de Buitrago J. 1995. La citometría de flujo en el laboratorio clínico.Sociedad Española de Bioquímica Clínica y Patología Molecular. Universidad de Salamanca. Pp: 37-39.
21) Orfao A, Gonzales M, San Miguel JF, López Borrasca A, L citometría de flujo en Hematología. En : " Libro de Symposia de la XXXI Reunión de las Asociación Española de Hematología y Hemoterapia". Torres Gómez A(ed) . Gráficas Milla, Córdoba, 1989 :187192.
22) Orfoa A, Ruiz-Argüelles A. Citometría de flujo y su aplicación en Hematología. In Enciclopedia Iberoamericana de Hematología. Tomo I. Ediciones Universidad de Salamanca, España.pp : 161-175.
23) Pandolfi F, Loughran TP, Starkemann G, et al. Clinical course and prognosis of the lymphoproliferative disease of granular lymphocytes : A multicenter study. Cancer 1990, 65 : 341.
24) Richards SJ, Scott CS. Human NK cells in health and disease : Clinical, functional, phenotypic and DNA genotypic characteristics. Leukemia and lymphoma 1982, 7 : 377-399.
25) Rosemblatt JD, Golde DW, Wachsman W el al. A second isolate of HTLV-11 associated with atypical hairy-cell leukernia. New England Journal of Medicine, 1986, 315 : 372374.
26) San Miguel JF, González M, Orfao A, López Borrasca A. Inmunopatología de leucemias y linfomas. Mta.Medicina Interna (Barcelona) 1988, 6 : 9-46.
27) Sausville EA, Eddy JL, Makuch RW el al . Histopathologic staging at initial diagnosis of mycosis fungoides and Sesary Syndrome. Am Intem Med 1988, 109 : 372-382.
28) Suchi T, Lennert K, Tu L-Y, Kikuchi M, Sato E, Stansfeld Ag, Feller AC : Histopathology and immunohistochemistry of periferical Tcell lymphomas : a proposal for their classification. Journal Clinical Pathol 1987, 40 995-1015.
29) Terstappen LWMM, Johnsen S, Segers Nolten IJM, Lohen MR. Identification and characterization of plasma cells in normal human bone marrow by high resolution flow cytometry. Blood 1990, 76 : 1739-1747.
30) Urbano-lspizua A, Matutes E, Villamor N, Sierra J, Pujades A, Reverter JC, Feliu E, Cervantes F, Vives-Corrons JL, Monseraat C. The value of detecting surface and cytoplasmic antigens in acute myeloid leukemia. British Journal of Haematology, 1992, SI: 178-183.
* Laboratorio Clínico,
Hospital Calderón Guardia
** Laboratorio Clínico,
Hospital San Vicente de Paúl