NEUMOLOGÍA
ENFERMEDAD PULMONAR
INTERSTICIAL DIFUSA
(Segunda Parte)
Vanessa Villalobos
Villalobos*, Alcibey Alvarado González**
Summary
The following
work on the subject is a review of information about the etiology and inmunopathogenesis,
still controversial, of idiopathic pulmonary fibrosis.
To attain
knowledge of the etiology, first, an allusion will be made on the roll that
different cells, mainly neutrophils and macrophages, play as mediators of
pulmonary parenchyma damage through different immunological mechanisms and
the roll of chemotactic factors, lymphokines and growth factors will be
emphasized.
Besides, based
on data found by electronic microscopy, the additional mechanisms responsable
of the interstitial thickening: partial or total alveolar collapse and apposition
of the alveolar septa, incorporation of intraalveolar exudates into the
alveolar septa; will be explained.
Last, because
of space reasons, only a short review on the pathogenesis of sarcoidosis
and pulmonary eosinophilia, will be made; because on further publication,
as continuation of this work, cases of interstitial pulmonary disease reports
some of these entities
Palabras claves:
ETIOLOGÍA
PATOGÉNESIS
FIBROSIS PULMONAR
INTERSTICIAL
SARCOIDOSIS
NEUMONÍA EOSINOFÍLICA
Key words
ETIOLOGY
PATHOGENESIS
INTERSTITIAL PULMONARY
FIBROSIS
SARCOIDOSIS
EOSINOPHILIC PNEUMONIA.
Resumen de Inmunopatogénesis
de Fibrosis Pulmonar:
Son varios
los motivos que han hecho pensar que la alveolitis fibrosante tiene una
base inmunológica, aunque el agente causal inicial no se haya establecido:
a) La presencia a menudo
de hiperglobulinemia, de factor reumatoide, de anticuerpos antinuclear
y de anticuerpos dirigidos en contra de los consituyentes pulmonares en
estos pacientes, inclusive en aquellos con aveolitis fibrosante no asociados
a enfermedades sistémicas.
b) La presencia de complejos
inmunes en el suero y ocasionalmente en la pared de los capilares pulmonares,
sobre todo en estadios tempranos, en aquellos con aveolitis fibrosante.
Macrófagos
Los macrófagos activados
secretan:
- Factores quimiotácticos
para neutrófilos.
- Factores que estimulan
la actividad secretora de fibroblastos y Neutrófilos.
- Proteasas y radicales
tóxicos-de oxígeno e hidroxilos al igual que los neutrófilos.
Mastocitos
Con la ayuda de tinciones
apropiadas se ha visto que estas células son numerosas en el tejido
intersticial y con la ayuda de la microscopia electrónica se ha demostrado
que los mastocitos se encuentran en aposición a los fibroblastos al
igual que las células epiteliales en regeneración, para proporcionar
estimulación adicional.
Linfocitos
La célula predominante
en el intersticio a pacientes con alveolitis fibrosante, es el linfocito,
el cual puede ser T colaborador (T4). como T supresor / citotóxico
(T8). Además, la Il-4 y la IL-5 predominan sugiriendo una
respuesta de tipo TH 2 (3), pese a que en otros estudios
la respuesta THI fue la vinculada con la aparición de inflamación
pulmonar progresiva llevando al paciente a fibrosis pulmonar. (1)
Células epiteliales
alveolares
Las células epiteliales
podrían ser reconocidas como autoantígenos por las células
T citotóxicos al mostrar expresión aberrante del antígeno
HLA-DR. Por otra parte, la proliferación epitelial regenerativa
vista al microscopio de luz como metaplasia cuboidal, común en
todas las etapas de la enfermedad, podría jugar un papel muy importante
en la patogénesis de esta entidad puesto que dicho epitelio libera
endotelina y factores fibrogénicos como el TNF-A y TGF-B.
Además, también se han descrito contactos directos entre
las células epiteliales y elementos del intersticio como los fibroblastos,
a través de deficiencias encontradas a lo largo de la membrana basal,
facilitándose de ésta manera el paso de cualquier mensaje
epitelial al tejido conectivo. La pérdida del epitelio y la
deficiencia de surfactante secundaria, puede ayudar a explicar el colapso
focal de los alveólos con aposición de las membranas basases
denudadas descrita en el síndrome de Hamman-Rich y el daño
alveolar difuso. La regeneración epitelial que cubre los orificios
de los alveólos colapsados, lleva a una pérdida permanente
de los alveólos, proceso de reparación pulmonar conocido como
induración atelectásica. Ocasionalmente, las membranas
basales epiteliales se cubren con fibrina en el daño alveolar difuso,
síndrome de Hamnan-Rich y durante las exacerbaciones de los casos
más crónicos. La incorporación de la fibrina
en el intersticio y su organización es otro proceso por el cual se
puede desarrollar fibrosis intersticial.
El endotelio capilar
El daño endotelial,
el edema intersticial y la fenestración del endotelio que se regenera
se observan en la microscopia electrónica. Dicho daño
endotelial promueve el edema intersticial al tornarse crónico y
contribuye a la fibrosis intersticial (8). La base autoinmune
de la UIP esta mejor establecida que para la DIP. La DIP probablemente
está más relacionada con la bronquiolitis respiratoria asociada
a enfermedad intersticial pulmonar vista en fumadores de cigarrillos y
considerada anteriormente.
Sarcoidosis
Patogénesis
La peculiar reacción
tisular granulomatosa propia de la Sarcoidosis sugiere que debe existir
un antígeno persistente, probablemente escasamente degradable que
inicie el proceso inmnológico activo, que puede resolverse espontáneamente
o progresar a la formación de granulomas, los cuales junto con la
alveolitis pueden causar daño al parénquima pulmonar y llevar
a la fibrosis pulmonar (1), (Fig. 1).
Después del estímulo antígenico se da activación
de los macrófagos locales y de las células T, por lo que van
a secretar IL1 e IL-2 respectivamente. La liberación de estos
mediadores tiene dos objetivos:
a) La estimulación
de la proliferación de células T a nivel local.
b) El reclutamiento de
células T provenientes de la sangre periférica.
El reclutamiento
de células T a nivel pulmonar podría explicar la Tlinfopenia
usualmente asociada a este desorden, y el probable gradiente de actividad
quimiotáctica de monocitos entre el pulmón y la sangre periférico.
El papel de las células T en la formación de la reacción
granulomatosa es de vital importancia puesto que éstas van a ser
las responsables de atraer al pulmón los monocitos provenientes de
la sangre periférica a través de la liberación del
factor quimiotáctico de monocitos, de la activación de estas
células a través de la liberación del interferón
-gamma, de la inmovilización de los macrófagos en la zona
de la inflamación a través de la liberación del factor
inhibitorio de la migración de los macrófagos y de la diferenciación
de las células B en células secretoras de inmunoglobulinas
(25), (Fig. 2). El macrófago
desempeña un papel clave en la inflamación crónica
debido a la gran cantidad de sustancias biológicamente activas que
puede producir, algunas de las cuales son tóxicas para las células.
Los estudios morfológicos de las biopsias de pulmón de macrófagos
involucrados especialmente en la formación de granulomas, sugieren
que estos están activos porque:
a. Se produce
un incremento en la capacidad de las células para liberar radicales
tóxicos de oxígeno así como enzimas microbicidas.
b. Los macrófagos
alveolares de pacientes con sarcoidosis activa liberan interleukina 1 de
manera espontánea.
c. Los macrófagos
alveolares de pacientes con sarcoidosis tienen una capacidad incrementada
de interactuar con el sistema de coagulación fibrinolítico.
Modulación
de la enfermedad en pacientes con sarcoidosis.
Linfocitos
T.
El proceso
de formación de granulomas está modulado por la presencia
de linfocitos T (10). Por ejemplo, en modelos animales
se ha demostrado que los linfocitos T colaboradores que tienden a ampliar
las respuestas celulares inmunes predominan en las etapas tempranas o de sarcoidosis
activa mientras que, los linfocitos T supresores que tienen un efecto opuesto
a los anteriores predominan en pacientes con enfermedad inactiva o estable.
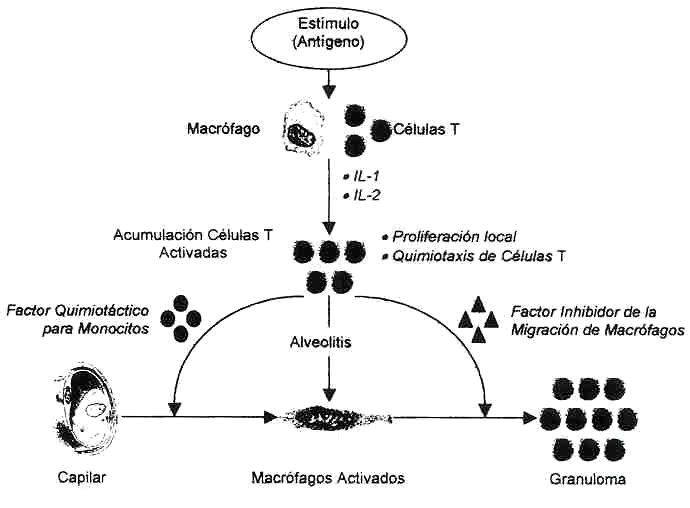
|
Fig.
1 Patogénesis de la formación del granuloma. Un estímulo
(el cual puede ser un antígeno) activa a los macrófagos para
liberar interleukina-1, y a las células-T para liberar interleukina-2.
La liberación de estos mediadores produce la acumulación
de células-T activas en el pulmón al estimular la proliferación
local de las mismas y la quimiotaxis de células-T sanguíneas.
Las células-T activadas que están presentes en el pulmón
regulan la formación del granuloma por medio de la secreción
de otras linfokinas, como el Factor Quimiotáctico para Monocitos
el cual atrae monocitos al pulmón. Otras linfokinas, como el
Factor inhibidor de la Migración de los Macrófagos y el interferón
inmune, estimulan la diferenciación de estas células en macrófagos
activados, los cuales forman una estructura compacta que forma el granuloma.
|
Secreción de
PGE2 por los macrófagos
Se ha demostrado
que la PGE2 inhibe la formación de granulomas, por lo que en sarcoidosis,
se postula que la capacidad de los macrófagos alveolares para liberar
la misma está marcadamente deprimida.
Fibrosis
Diversos estudios sugieren
que la IL -1 y el interferón desempeñan un papel importante
en la aparición del proceso fibrótico, puesto que actúan
como factores de crecimiento para fibroblastos y puesto que actúan
de manera sinérgica para incrementar la proliferación de
los mismos. Sin embargo también existen otros factores tales
como la fibronectina y un factor de crecimiento derivado de los macrófagos,
que dan lugar a la proliferación de fibroblastos y al depósito
de colágeno.
Producción
de Inmunoglobulinas
La hipergamaglobulinemia
se encuentra frecuentemente en pacientes con sarcoidosis, lo que sugiere
que probablemente los eventos que llevan a la formación de granulomas
llevan a la producción de inmunoglobulinas. Para esto se requiere
que los macrófagos presenten antígenos a los linfocitos T
y que éstos liberen IL-1. Así, las células T
activadas de esta manera, liberarán una variedad de linfocinas importantes
en la formación de la alveolitis y de los granulomas y linfocinas
adicionales como el factor de crecimiento de células B y el factor
de diferenciación de células B importantes para estimular
la producción de inmunoglobulinas por las células plasmáticas.
(25).
Algunas
de las inmunoglobulinas producidas probablemente estén dirigidos
contra un antígeno específico mientras que otras inmunoglobulinas
probablemente estén dirigidos contra múltiples antígenos
(10, 25).
Eosinofilia pulmonar
Conceptos generales
El eosinófilo
juega un papel importante en la patogénesis de las enfermedades pulmonares
eosinofilicas (20). Los eosinófilos son producidos
en la médula ósea bajo el control de citocinas secretadas
por los linfocitos T colaboradores tipo 2, principalmente la IL-5, que juega
un rol central en la maduración, activación y sobrevivencia
de los eosinófilos. Una vez liberados al torrente sanguíneo,
permanecerán en éste de 12 a 18 h en sujetos normales y
más tiempo en ciertos estados de enfermedad (16).
Normalmente la sangre contiene de 50 a 250 eosinófilos células/uL.
Es importante recalcar que el conteo absoluto de eosinófilos sanguíneos
es más importante que el porcentaje de los mismos. En este
aspecto es importante tener presente que la eosinofilia tisular no necesariamente
se correlaciona con la eosinofilia sanguínea (aunque en la mayoría
de los casos ambas se encuentran asociadas), ya que, los conteos de eosinófilos
sanguíneos, pueden descender a medida que éstos empiezan
a invadir el pulmón y luego más bien aumentar a medida que
el infiltrado se resuelve, debido a un retardo en la respuesta de la médula
ósea (12).
b- Esputo
y LBA
Los conteos de eosinófilos
en el esputo y en el LBA reflejan mejor la eosinofilia tisular que los
conteos de estos en sangre periférico, pero también, los eosinófilos
al igual que los neutrófilos, son lavados más fácilmente
del pulmón que los macrófagos, dando como resultado que los
conteos de los mismos se encuentren elevados en enfermedades como la alveolitis
fibrosante, entidad que raramente está caracterizada por un grado
apreciable de eosinofilia tisular. El valor normal de eosinófilos
en el LBA, usualmente expresado como porcentaje del total de células
recuperadas del pulmón es de menos del 3%. Por otra parte, los
eosinófilos en el esputo son generalmente expresados en términos
cualitativos.
c- Biopsia:
La biopsia es la única
manera de establecer a ciencia cierta el diagnóstico de eosinofilia
tisular, sin embargo, en la práctica común, las características
del cuadro clínico, los hallazgos en el hemograma, esputo y LBA
(difícil de evaluar en nuestro medio), son suficientes para establecer
el diagnóstico. En caso de ser necesaria la biopsia, bastará
con realizar una biopsia, transbronquial puesto que en las eosinofilias
pulmonares las áreas de pulmón involucradas en el proceso
son lo suficientemente grandes para ser abordadas con éxito por el
broncoscopista. Sin embargo, algunas eosinofilias pulmonares, caracterizados
por la presencia de vasculitis, se hacen acreedores a una biopsia por toracoscopia,
debido a que las vasculitis raramente pueden ser diagnosticadas por medio
de una biopsia transbronquial por problemas de tamaño de la muestra
(18).
Papel
de los eosinórilos:
Los eosinófilos
penetran en los tejidos bajo la influencia de agentes quimiotácticos
producidos por varias células (particularmente los mastocitos).
Estas células secretan una variedad de sustancias, algunas de las
cuales contrarrestan la acción de sustancias secretadas por los mastocitos;
por ejemplo, la histaminasa y la aril sulfatasa destruyen la histamina y
la sustancia de reacción lenta de anafilaxis secretadas por los mastocitos,
respectivamente. Además, los eosinófilos fagocitan complejos
inmunes y gránulos de los mastocitos y tienden a amortiguar reacciones
de hipersensibilidad. Pero, a pesar de esas acciones, también
secretan sustancias como la proteína básica mayor y la proteína
canónica eosinofílica que pueden ser tóxicas para las
células del húesped, por lo que se piensa, pueden ser responsables
del daño tisular observado en los desórdenes eosinofílicos.
En la neumonía eosinofilia criptogénica la demostración
de expresión de moléculas de adhesión intercelular
por éstas células, sugiere que las mismas han sido activadas,
probablemente bajo la influencia de Linfocitos T activados. Por otra
parte, el hecho de que la microscopio electrónica haya mostrado la
liberación de gránulos eosinofílicos en áreas
cercanas a el tejido alveolar degenerado y necrótica, se sugiere que
ésta célula juega el papel central en el daño tisular
observado en los pacientes con neumonías eosinofílicas (4).

|
|
Fig
2. Mecanismos de producción de inmunoglobulinas. Gracias
a la acumulación de células-T activadas en el pulmón
se liberan linfocinas adicionales: El Factor de crecimiento de células-B
(BCGF) y el Factor de diferenciación de células-B (BCDF).
Estos factores estimulan a la scélulas-B para que produzcan inmunoglobulinas.
Algunas de las inmunoglobulinas producidas de esta manera están
dirigidas contra un antígeno específico, mientras que otra
porción de inmunoglobulinas es policional (dirigidas a múltiples
antígenos) V no están directamente relacionadas con la enfermedad
subyacente.
|
Resumen
La presente
exposición del tema es una recopilación de información
acerca de la etiología e inmunopatogénesis aún controver-sial
de la fibrosis pulmonar idiopática. Para lograr el conocimiento
de la misma primeramente se hará una alusión al rol que
juegan las diferentes células, principalmente neutrófilos
y macrófagos, como mediadores del daño al parénquima
pulmonar a través de los diferentes mecanismos inmunológicos
y se enfatizará el papel desempeñado por diversos factores
quimiotácticos, linfocinas y factores de crecimiento. Además,
basados en los datos puestos de manifiesto por la microscopio electrónica
se explicarán los mecanismos adicionales responsables del engrosamiento
del intersticio como el colapso parcial o total de los alvéolos y
la aposición de los septos alveorales, así como, la incorporación
de los exudados intraalveolares dentro de los septos alveolares. Por
último, se hará una breve revisión por razones de
espacio, de la patogénesis de la sarcoidosis y de la eosinofilia
pulmonar, ya que en una casuística de enfermedades pulmonares intersticiales
próxima a publicarse como continuación del presente trabajo,
se reportan algunos casos de dichas entidades.
Bibliografía
- Antoniades,
H.N. et al. Platelet-derive Growth Factor in Idiopathic Pulmonary
Fibrosis. J. Clin. Invest. 1990; 86: 1055-1064.
- Aston,
C. et al. Enhanced Insulin-like Growth Factor Molecules in Idiopathic
Pulmonary Fibrosis. Am. J. Respir. Crit. Care
Med. 1995; 151:1597-1603.
- Baumagartnr,
K. B. et al. Cigarette Smoking: Arisk Factor for Idiopathic Pulmonary
Fibrosis. Am. J. Respir. Crit. Care Med. 1997;
155: 242-248.
- Castella,
J. et al. Lavado broncoalveolar. Arch, Bronconeumol. 1997;
33: 515-526.
- Castella,
J.; Ancochea, J., Llorente, L; Puzzo, C.; Sanchis, J.; Suerio, A.; Xaubet,
A. Lavado broncoalveolar. Normativas Separ. Arch. Bronconeumol.
1997; 33: 530-542.
- Corrin,
B. Diseases characterized by restrictive ventilatory impairment.
In: Pathology of the Lung. London. Churchill-Livingstone. 2000.
(Ist. Ed.): 239-277.
- Corrin,
B. Pathology of Interstitial Lung Disease. Seminars in Respiratory
and Critical Care Med. 1994; 15: 61-76.
- Crofton,
J. W.; Livingstone, J.L.; Oswald, N. C., et al: Pulmonary eosinophilia.
Thorax. 1952-, 7: 1.
- Coxson,
H. Q., et al. Quantification of Idiopathic Pulmonary Fibrosis using
compute Tomography and Histology. Am, J. Respir. Crit.
Care Med. 1997; 155: 1649-1656.
- Chapman,
H. A.; Allen, C. L.; Stone, 0. L. Abnormalities in Pathways of Alveolar
Fibrin turn over among Patients with Interstitial Lung Disease. Am.
Rev. Respir. Dis. 1986; 133: 437-44.
- Enright,
T.; Chau, S.; Lim, D.T. Pulmonary eosinophilic syndromes. Ann.
Allergy. 1989; 62: 277.
- Fraser,
S. R.; Mueller, L.N.; Colman, N.; Paré, D. P. Interstitial
Pneumonitis and Fibrosis. Diagnosis of Diseases of the Chest.
Philadelphia. W. B. Saunders Co. 1999 ( 4th Ed. ): 1584-1626.
- García,
J. G. N.; Wolvwn, R. García, P.L.; Keogh, B. A. Assessment of Interlobar
Variation of Bronchoalveolar Lavage Cellular Differentials In Interstitil
Lung Diseases. Am. Rev. Respir. Dis. 1986; 133:
444-449.
- Glich,
G. J. Cuffent understanding of eosinophil function. Hospital Practice.
1988; 23: 137-160.
- Guido,
M. 1. Alvarado, G. A. La reacción tardía en asma bronquial.
A.M.C. 1991; 34: 94-111.
- Hunninghake,
G. W., et al. Mechanisms of Neutrophil Accumulation in the Lungs
of Patients with Idiopathic Pulmonary Fibrosis. J. Clin . Invest.
1981; 68: 259-269.
- Hunninghake,
G. W.; Fulmer, J. D.; Young. R. C. Jr.; Gadex, J. C. Cristal, R.
G. Localization ot the Immune Response in sarcoidosis. Am. Rev.
Respir. Dis. 1979; 126: 49-57.
- Jederlinic,
P. C.; Sicilian, L.; Gaensier, E.A. Chronic eosinophilic pneumonia:
a report of 19 cases and review of the literatura. Medicine. (Baltimore)
1988; 67: 154-162.
- Katzenstein,
A. A.; Myers, J. L. Nonspecific Interstitial Pneumonia Classification
and Diagnostic Criteria. Am. J. Surg. Path. 2000; 24: 1-3.
- Katzenstein,
A. A. et al. Pathogenesis of "Fibrosis" in Interstitial Pneumonia:
An Electron Microscopic Study. Hum, Pathol. 1985; 16: 1015-1024.
- Keogh,
A. B.; Bernardo, J.; Hunninghake, W. G.; Line, R. B.; Price, L. D.; Crystal,
G. R. Effectf of intermittent High Dose Parenteral Corticosteroid on the
alveolitis of idiopathic pulmonary fibrosis. Am. Rev. Respir.
Dis. 1983; 127: 18-22.
- Kuhn,
C, et al. An Immunohistochemical Study of Architectural Remodeling
and Tissue Synthesis in Pulmonary Fibrosis. Am. Rev.
Respir. Dis. 1989; 140: 1693-1703.
- Schwartz,
D. A. et al. Determinants of Progression in Idiopathic Pulmonary
Fibrosis. Am. J. Respir. Crit. Care Med. 1994; 149:
444-449.
- Snider,
L.G. Onterstitial Pulmonary Fibrosis-Which Cell is the Culprit ?. Am Rev.
Respir. Dis. 1983; 127: 535-539.
- Thomas,
P.D.; Hunninghake, G. W. Current Concepts of the Pathogenesis of Sarcoidosis.
Am, Rev. Respir. Dis. 1987; 135: 747-760.
- Ward,
P.; Hunninghake, G. W. Lung Inflammation and Fibrosis. Am.
J. Respir. Crit. Care Med. 1998; 157: 123-129.
* Médico Interno. Escuela de
Medicina. Universidad de Costa Rica . HOSPITAL SAN JUAN DE DIOS.
2001
** Internista y Neumólogo. Laboratorio de
Fisiopatología Respiratoria. Servicio de Neumología.
HOSPITAL SAN JUAN DE DIOS. SAN JOSE. COSTA RICA. Member
of American Association for Respiratory Care. U.S.A .